
Weekly reads 8/06/26
The invisible foundations of cancer: early malignancy, epigenetic plasticity, and the tools to see it all

This week’s reads illuminate the hidden architectures of early malignancy, the epigenetic chaos unleashed by chromosomal loss, and the cutting-edge tools reshaping how we see and interpret cellular life. In precursors of squamous cell carcinoma, a distinct population of malignant keratinocytes maintained by a ΔNp63/PITX1 axis demonstrates that pre-invasive tumors have already acquired genomic and transcriptional features of aggressiveness, whereas the invasion mechanism is not developed until later stages. At the same time, loss of the Y chromosome in lung adenocarcinoma changes the epigenetic landscape in such a way that it gives rise to EMT, lineage plasticity, and increased metastatic potential, thereby showing that chromosomal loss can be just as transformative as an oncogenic mutation. On the technology side: MetaboRamics uses 16-plex Raman imaging for live-cell spatial metabolomics, revealing the metabolic rewiring dynamics of EMT and stress responses, CytoSignal identifies ligand-receptor interactions at the cellular level, confirming its predictions through proximity ligation assays. Further, in the field of data and models, a bold research overturns the "bigger is better" belief for single-cell foundation models, indicating that performance reaches a plateau even with only 1, 10% of current datasets, which may mean that quality, rather than quantity, is the secret to fully realizing their potential.
Preprints/articles that I managed to read this week
p63 and PITX1 sustain a pre-invasive malignant keratinocyte population in squamous cell carcinoma precursors
Staeger et al. bioRxiv (2026). 10.64898/2026.05.21.725073
The paper in one sentence
A discrete population of malignant keratinocytes (ASK) in actinic keratosis harbors UV-associated mutations and copy number alterations, is sustained by a ΔNp63/PITX1 regulatory axis that blocks differentiation, and shares core oncogenic programs with invasive squamous cell carcinoma while lacking invasion effectors.
Summary
Actinic keratosis (AK) is a common precursor to cutaneous squamous cell carcinoma (cSCC), but the cellular identity and molecular programs of early malignant keratinocytes have been poorly defined. The authors performed CITE-seq (simultaneous transcriptome and surface proteome) on patient-matched AK, UV‑exposed normal skin, and non‑UV‑exposed normal skin (n=5 patients, 12 biopsies) and spatial whole‑transcriptome profiling in an independent cohort (n=4). They identify AK‑specific keratinocytes (ASK), a discrete population enriched in dysplastic basal epidermis. ASK show genomic hallmarks of malignancy: dominant UV‑associated SBS7b mutational signature, high mutational burden (median 20.75 muts/Mb), recurrent copy number alterations (9p loss harboring CDKN2A, 8q gain harboring MYC), and TP53 overexpression with reduced p53 pathway activity. Transcriptomically, ASK occupy a basal‑like, undifferentiated state, characterized by upregulation of ΔNp63 and PITX1, downregulation of Notch/HES1 signaling, and activation of glycolytic metabolism (SLC2A1/GLUT1). Comparison with published cSCC data reveals that ASK share core oncogenic programs with invasive tumor‑specific keratinocytes (TSK) – including IGFBP6, IGFBP2, ITGA6 – but lack invasion‑associated effectors (MMP1, MMP10, PTHLH). IGFBP6 is validated as a pro‑proliferative factor in the AK‑derived PM1 cell line (knockout reduces proliferation, recombinant protein increases it). The AK microenvironment shows expansion of inflammatory basal keratinocytes, barrier disruption, and early immunosuppressive T cell remodeling (CD8⁺ exhausted T cells, Tregs, Th17‑skewed populations). The study proposes that ASK represent the earliest malignant keratinocyte population, sustained by ΔNp63/PITX1, and that core dependencies established at this pre‑invasive stage may be retained in invasive tumors, offering potential targets for prevention or treatment.
Personal highlights
Genomically defined malignant population in a pre‑invasive lesion: ASK are enriched in dysplastic epidermis and exhibit definitive malignant hallmarks: UV‑dominant SBS7b mutational signature (27.1% contribution), high mutational load (significantly higher than other keratinocyte clusters), recurrent 9p loss (CDKN2A) and 8q gain (MYC), and TP53 overexpression with reduced p53 signaling – placing them on the trajectory from normal skin to invasive cSCC.
ΔNp63/PITX1 regulatory module maintains an undifferentiated state: ASK show selective regulon activity for TP63 and PITX1, with preferential expression of the ΔNp63 isoform (which maintains epithelial stemness). This axis attenuates Notch/HES1‑driven differentiation (lowest NOTCH hallmark score) and activates glycolytic metabolism (SLC2A1 upregulation, glycolysis pathway enrichment), consistent with p63‑driven metabolic reprogramming in squamous cancers.
ASK share core oncogenic programs with invasive cSCC but lack invasion effectors: Overlap analysis between ASK and published tumor‑specific keratinocyte (TSK) signatures reveals nine shared genes (IGFBP6, IGFBP2, ITGA6, PKM, LAMA3, etc.) representing early dependencies. Crucially, canonical TSK invasion markers (MMP1, MMP10, PTHLH) are not elevated in ASK, distinguishing early malignant programs from later invasive effectors.
IGFBP6 as a pro‑proliferative effector in pre‑malignant keratinocytes: IGFBP6 is among the most upregulated ASK markers, confirmed at protein level in AK tissue. Knockout of IGFBP6 in the AK‑derived PM1 cell line reduces proliferation, while recombinant IGFBP6 increases it, nominating IGFBP6 as a candidate driver of clonal expansion in early squamous carcinogenesis.
Early immune remodeling in the AK microenvironment: AK lesions show expansion of an inflammatory basal keratinocyte state (IL20, CCL2, CXCL2), depletion of terminally differentiated granular cells, and increased T cell infiltration with exhausted CD8⁺ T cells (exclusive to AK), Tregs, and Th17‑skewed populations with oligoclonal TCR expansions, indicating that immunosuppressive circuits are already established at the pre‑invasive stage.
Why should we care?
This study offers a valuable resource and a clear conceptual framework: early malignant cells in squamous carcinogenesis are already transcriptionally distinct, sustained by a ΔNp63/PITX1 circuit, and share core dependencies with invasive tumors. This suggests that targeting these early programs might prevent progression rather than merely treating established cancers. But the path from this descriptive atlas to clinical application is long, and the work should be viewed as hypothesis‑generating. Rigorous functional studies in animal models, larger validation cohorts, and prospective clinical trials will be required before any of these candidates can be considered for prevention or therapy.
Loss of the Y chromosome drives epigenetic and transcriptomic plasticity in lung adenocarcinoma
Schlüter K. et al., bioRxiv (2026). DOI: 10.64898/2026.06.01.729186
The paper in one sentence
This study demonstrates that loss of the Y chromosome (LOY) in lung adenocarcinoma (LUAD) drives epigenetic reprogramming, lineage plasticity, and metastatic potential by triggering epithelial-to-mesenchymal transition (EMT) and increasing cellular adaptability to stress.
Summary
Using multi-omic profiling (whole-genome sequencing, single-cell RNA-seq, proteomics, and epigenetic assays) of primary LUAD samples and isogenic A549 cell models, the authors show that LOY is prevalent in malignant cells and associated with poor clinical outcomes. Mechanistically, LOY causes haploinsufficiency of Y-linked dosage-sensitive regulators, leading to DNA hypomethylation at EMT gene promoters (e.g., THY1, LOX) and H3K4me3 enrichment, which collectively destabilize the chromatin landscape. This epigenetic remodeling induces EMT, inflammatory signaling, and stemness features, while increasing cell-to-cell heterogeneity and lineage plasticity. Functionally, LOY does not confer a proliferative advantage under basal conditions but enhances clonogenic potential, resilience to metabolic stress (glucose/glutamine deprivation), and resistance to genotoxic stress (ionizing radiation). In vivo, LOY cells exhibit a selective advantage during tumor engraftment and metastatic dissemination, with LOY clones disproportionately contributing to metastasis in xenograft models. Clinically, LOY correlates with aggressive disease, increased metastasis, and shorter overall survival in male LUAD patients.
Personal highlights
LOY as a driver of EMT and plasticity: LOY triggers EMT programs (e.g., THY1, LOX upregulation) and increases lineage plasticity, enabling rapid adaptation to stress and metabolic challenges.
Epigenetic remodeling: LOY induces focal DNA hypomethylation and H3K4me3 enrichment at EMT gene promoters, destabilizing chromatin and amplifying transcriptional heterogeneity.
Functional resilience: LOY cells show enhanced clonogenic capacity and resistance to metabolic (glucose/glutamine deprivation) and genotoxic stress (radiation), without baseline proliferative advantages.
Metastatic advantage: In vivo models reveal that LOY cells are selectively enriched in metastases, with LOY clones exhibiting a 21% higher metastatic capacity than ROY clones.
Clinical relevance: LOY is associated with poor survival in male LUAD patients, with low Y-linked gene expression predicting shorter overall survival (median 50 vs. 122 months).
Why should we care?
This work reframes loss of the Y chromosome (LOY), previously dismissed as a neutral byproduct of genomic instability, as a key driver of tumor evolution in lung adenocarcinoma. The main takeaway is that LOY acts as an epigenetic "gatekeeper": its loss destabilizes cellular identity, enabling tumors to adapt, evade treatment, and metastasize more effectively. This challenges the traditional view of cancer progression as solely driven by oncogenic mutations, instead highlighting how chromosomal loss can rewrite the epigenetic landscape to fuel aggression.
MetaboRamics: Highly multiplexed metabolic imaging by stimulated Raman for spatial metabolomics in live cells
Chadha R.S. et al., bioRxiv (2026). DOI: 10.64898/2026.05.21.727012
The paper in one sentence
This study introduces MetaboRamics, a 16-plex stimulated Raman scattering (SRS) microscopy platform that enables live-cell spatial metabolomics, revealing dynamic metabolic rewiring during processes like epithelial-to-mesenchymal transition (EMT) and cellular stress responses.
Summary
The authors developed MetaboRamics, a super-multiplexed SRS imaging platform that combines nine bioorthogonal Raman probes (targeting glucose uptake, lipid synthesis, choline metabolism, DNA synthesis, amino acid incorporation, and organelle markers) with five label-free channels (proteins, lipids, unsaturated lipids, saturated triglycerides) and two autofluorescence channels (NADH, FAD) for redox state readouts. This 16-plex system allows simultaneous, non-destructive, and spatially resolved tracking of multiple metabolic pathways in live cells. The platform was validated by profiling EMT in A549 cells, revealing a global attenuation of metabolic activity in mesenchymal cells, including reduced glucose-derived biomass, lipid turnover, protein synthesis, and altered redox balance. Additionally, MetaboRamics was used to phenotype cellular responses to nine metabolic stressors (e.g., fructose overload, palmitic acid, serum deprivation, inflammation, and pharmacological perturbations), uncovering pathway-specific adaptations and subcellular metabolic heterogeneity. For example, inflammatory stimuli and EMT both induced global metabolic downregulation, while pharmacological inhibitors (e.g., CHX, MG132) revealed distinct mechanisms of action on protein synthesis and glucose metabolism.
Personal highlights
16-plex live-cell metabolic imaging: Integration of nine bioorthogonal probes (e.g., EdU for DNA synthesis, d₇-glucose for lipid/protein synthesis, d₈-arachidonic acid for PUFA uptake) with five label-free channels and TPEF redox imaging, enabling unprecedented multiplexing for spatial metabolomics.
Metabolic rewiring in EMT: Mesenchymal cells exhibit ~30% reduction in glucose-derived biomass, lipid turnover, and protein synthesis, with decreased mitochondrial activity and NADH levels, indicating a globally attenuated metabolic state.
High-throughput stress phenotyping: Systematic profiling of nine metabolic stressors (e.g., fructose, palmitic acid, inflammation, drug treatments) revealed distinct and sometimes counterintuitive metabolic responses, such as increased PUFA uptake under palmitic acid stress (a potential lipotoxicity rescue mechanism).
Subcellular resolution: Spatial segmentation (cytoplasm, nucleus, nucleoli) uncovered compartment-specific metabolic changes, e.g., nucleoli-specific protein synthesis attenuation under inflammatory stress.
Technical robustness: Minimal photodamage, high signal-to-noise ratios, and ~25-minute acquisition time for 16-plex imaging, with validation via single-channel controls and orthogonal methods (e.g., TPEF, spontaneous Raman).
Why should we care?
MetaboRamics addresses an important gap in live-cell metabolomics: the ability to simultaneously visualize multiple metabolic pathways in real time, at subcellular resolution, and without destroying the sample. For non-scientists, the takeaway is that this platform reveals how cells dynamically rewire their metabolism, not just globally, but in specific organelles and under diverse stressors, providing a window into the hidden metabolic heterogeneity that drives disease progression, drug resistance, and cellular adaptation. While MetaboRamics is a major leap forward, it has limitations. The spectral overlap of Raman tags restricts the palette to 16 channels, and laser tuning speed limits throughput. Sensitivity is also constrained to the low μM–mM range, missing many low-abundance metabolites. Future improvements, such as faster lasers, epr-SRS for enhanced sensitivity, or AI-assisted unmixing, could expand its capabilities. Additionally, the biocompatibility and potential perturbation of bioorthogonal probes (despite optimization) warrant further validation in primary cells and in vivo.
Many Needles in a Haystack: Active Hit Discovery for Perturbation Experiments
Rubbi et al. arxiv (2026). https://arxiv.org/abs/2605.10196
The paper in one sentence
This work introduces Probability-of-Hit, an acquisition function for active learning that directly maximizes the discovery of threshold-exceeding perturbations in high-throughput experiments, improving hit recovery by up to 6.4% over baselines on real biological datasets.
Summary
High-throughput gene perturbation technologies (e.g., CRISPR screens) enable parallel testing of thousands of genetic interventions, but experimental budgets remain severely limited. The core challenge is hit discovery: identifying as many perturbations as possible whose phenotypic effect exceeds a predefined threshold, rather than locating a single global optimum. Traditional approaches are misaligned with this goal. Pure exploration wastes budget on low-value regions, while optimization methods over-exploit dominant modes and neglect disconnected high-response areas, performing poorly in the multimodal, heterogeneous landscapes typical of biological systems. The authors formalize hit discovery as a closed-loop experimental design problem and propose Probability-of-Hit (PoH), an acquisition function that ranks candidates by their posterior probability of exceeding the hit threshold. This directly targets the discovery objective, balancing exploitation of known high-response regions with exploration of promising but uncertain candidates. They prove asymptotic optimality of PoH under standard assumptions (posterior concentration, margin conditions) and demonstrate its effectiveness across synthetic benchmarks and five real-world CRISPR screening datasets (e.g., Schmidt IL-2, Zhuang NK). Empirically, PoH consistently outperforms baselines, recovering more hits under fixed budgets. For example, on the Schmidt IL-2 dataset, PoH achieves a 6.4% improvement in cumulative hit ratio over the next best method.
Personal highlights
Formalization of hit discovery as a distinct objective: The paper clarifies that hit discovery (maximizing the number of threshold-exceeding perturbations) differs fundamentally from optimization or pure exploration, requiring tailored acquisition strategies for multimodal biological landscapes.
Probability-of-Hit acquisition function: A novel, principled approach that ranks candidates by their posterior probability of being a hit (p(f(g)>τ) directly aligning acquisition with the discovery goal rather than surrogate objectives like uncertainty reduction or global optimization.
Theoretical guarantees: Proof of asymptotic optimality for PoH, showing it recovers at least Tb/2−o(Tb) hits with high probability under standard assumptions, where TTT is the number of rounds and bbb is the batch size.
Empirical validation on real biological data: Outperforms state-of-the-art baselines across five large-scale CRISPR screening datasets (e.g., Schmidt IL-2, Zhuang NK), with gains of up to 6.4% in cumulative hit ratio, demonstrating robustness in complex, high-dimensional settings.
Batch-aware design for high-throughput experiments: The algorithm supports parallel evaluation of multiple perturbations per cycle, a critical feature for modern pooled screens, and reveals that increasing batch size yields larger gains than refining hit thresholds.
Evaluating the role of pretraining dataset size and diversity on single-cell foundation model performance
DenAdel et al. Nature Methods (2026). 10.1038/s41592-026-03120-y
The paper in one sentence
This large-scale study reveals that single-cell foundation models (scFMs) plateau in performance with only a small fraction (1–10%) of current pretraining datasets, challenging the assumption that “bigger data always yields better models” in single-cell biology.
Summary
The success of transformer-based foundation models in NLP and computer vision has inspired analogous efforts in single-cell biology, where models like scBERT, Geneformer, and scGPT are trained on tens of millions of cells. However, the scaling laws that govern performance in these domains, where larger datasets consistently improve model capabilities, remain unproven for single-cell transcriptomics. This study addresses this gap by pretraining 400 models on subsets of the 22.2-million-cell scTab corpus and evaluating them across 6,400 experiments spanning zero-shot and fine-tuned tasks, including cell-type classification, batch integration, and perturbation response prediction. The authors tested five model architectures (PCA, scVI, SSL, Geneformer, SCimilarity) and three downsampling schemes (random, cell-type re-weighted, geometric sketching) to assess the impact of dataset size (1–100%) and diversity on performance. Surprisingly, they found that most models saturated at 1–10% of the full dataset (as little as ~200,000 cells), with no clear performance gains from larger or more diverse pretraining corpora. Even spiking in perturbation data (e.g., from Perturb-seq) failed to improve downstream task performance. While larger models (e.g., higher parameter counts) tended to perform better in absolute terms, they still plateaued with small datasets, and simple baselines (e.g., PCA, scVI) often outperformed transformer-based models like Geneformer. These results suggest that current scFMs are not data-limited and that scaling datasets further may not yield meaningful improvements.
Personal highlights
Performance saturation at small dataset sizes: Across all tasks (cell-type classification, batch integration, perturbation response prediction), models typically reached 95% of peak performance at 1–10% of the 22.2M-cell corpus, with 1% (~200K cells) often sufficient for near-optimal results.
No evidence of LLM-like scaling laws: Unlike large language models, single-cell foundation models do not show consistent performance improvements with increased pretraining data size, indicating that blindly scaling datasets may not be an effective strategy.
Dataset diversity does not improve performance: Neither cell-type re-weighting (balancing cell-type proportions) nor geometric sketching (sampling uniformly across transcriptional space) outperformed random downsampling, suggesting current diversity strategies are ineffective for scFMs.
Simple baselines outperform transformers: Pretrained PCA and scVI often matched or exceeded the performance of more complex models (e.g., Geneformer, SSL) on classification and batch integration tasks, raising questions about the need for transformer architectures in single-cell applications.
Model size helps, but with diminishing returns: Larger models (e.g., higher parameter counts) improved absolute performance but still saturated with small datasets, and gains diminished with each successive increase in scale.
Why should we care?
This paper provides an urgently needed dose of reality regarding scFMs: more data does not necessarily imply a more accurate model. While in NLP scaling rules can accurately predict the improvement with more data, scFMs seem to converge very early, needing just a minimal amount (1–10%) of existing training sets to achieve nearly optimal accuracy. For those outside the field, what needs to be emphasized is that the common “the more data, the better model” approach cannot always be applied; in the realm of single-cell research, high-quality data, proper alignment of tasks, and efficient modeling can play much more important roles. In terms of practical applications, the results of the study provide food for thought for both researchers and funders. Instead of racing to build ever-larger models, the field should prioritize:
Data curation: Focusing on high-quality, task-relevant datasets rather than indiscriminate scaling.
Task alignment: Ensuring pretraining objectives (e.g., masked gene prediction) match downstream applications (e.g., cell-type annotation).
Model efficiency: Exploring architectures beyond transformers, as simpler models (PCA, scVI) often perform just as well.
Nonetheless, there are several key limitations of the study. Only five architectures were tested and a subset of tasks (classification, integration, perturbation prediction) was used for evaluation, which does not guarantee that all possible use cases where scFMs could shine were considered. Furthermore, the fact that perturbation spikes did not prove useful indicates the misalignment of currently applied pretraining techniques with the hardest problems in biology. Also, there is a lack of experiments with compute-optimal scaling (the balance between model parameters, training data, and compute), which is known to lead to breakthrough advances in NLP. Finally, one should note that simple representations such as PCA and HVGs perform better than advanced models, indicating the intrinsic weakness of “cell as a sentence of genes” approach.
Depth normalization for single-cell genomics count data
Booeshaghi et al. bioRxiv (2026). 10.1101/2022.05.06.490859
The paper in one sentence
This work introduces PFlogPF (proportional fitting-log-proportional fitting), a mathematically principled normalization method that uniquely satisfies variance stabilization, depth normalization, and monotonicity—three critical but often conflicting desiderata for single-cell RNA-seq count data.
Summary
Normalization of single-cell RNA-seq counts is a foundational step that shapes all downstream analyses, yet existing methods often fail to simultaneously achieve variance stabilization (for PCA/clustering), depth normalization (to remove sequencing depth bias), and monotonicity (to preserve within-cell gene rankings). The authors prove that PFlogPF, a two-step proportional fitting approach bracketing a log transformation, is the only feature-relabeling-equivariant method that satisfies all three properties. PFlogPF is mathematically equivalent to the shifted centered log-ratio (CLR) transform, a compositional data method developed over 40 years ago but underutilized in single-cell genomics. The study benchmarks 8 normalization methods (including sctransform, log1pPF, CPM, and Seurat/Scanpy defaults) across 526 datasets (437 passing QC), demonstrating that PFlogPF consistently outperforms alternatives. It eliminates residual depth structure while preserving variance stabilization and monotonicity, unlike log1pPF (which reintroduces depth bias after logging) or sctransform (which scrambles gene rankings). Critically, PFlogPF shows superior robustness to downsampling, recovering 36.8/50 nearest neighbors on average in k-NN graphs after depth reduction, compared to just 5.8/50 for other methods. It also exhibits greater stability to feature-panel choice, with higher Jaccard overlap (0.756 vs. 0.677 for log1pPF) when switching between gene sets. The authors further reveal that Seurat’s “CLR” implementation does not perform true CLR, highlighting a widespread mislabeling in popular workflows.
Personal highlights
Theoretical optimality: PFlogPF is the only normalization method that simultaneously achieves variance stabilization, depth normalization, and monotonicity while being invariant to feature relabeling.
Equivalence to shifted CLR: PFlogPF is mathematically identical to the shifted centered log-ratio transform, linking single-cell genomics to decades of compositional data theory and resolving a long-standing gap in the field.
Unmatched benchmark performance: Across 526 datasets, PFlogPF outperforms 7 alternatives (including sctransform, log1pPF, CPM) on all three key criteria, with no residual depth correlation and perfect rank preservation within cells.
Robustness to downsampling: PFlogPF preserves local neighborhood structure in k-NN graphs even after artificial depth reduction, recovering 36.8/50 neighbors vs. 5.8/50 for other methods, a critical advantage for real-world data with variable sequencing depth.
Stability to feature selection: Unlike log1pPF, PFlogPF’s induced geometry is insensitive to gene panel choices, reducing avoidable variation in large-scale analyses and foundation model training where feature sets may vary.
Why should we care?
This paper addresses a deceptively critical but overlooked problem: normalization is the invisible foundation of single-cell analysis, and flaws here cascade into erroneous biological conclusions. Many researchers treat normalization as a “black box” preprocessing step, but the authors show that common defaults (e.g., log1pPF, sctransform) fail to fully remove depth bias or preserve gene rankings, leading to spurious differential expression calls, distorted cell-cell distances, and misinterpreted markers. The key insight is that a second proportional fitting step after the logarithm (PFlogPF) elegantly solves these issues. For non-specialists, the takeaway is that not all normalization methods are equal, PFlogPF provides a principled, one-size-fits-most solution that addresses the core pitfalls of current approaches. Its equivalence to the shifted CLR transform also connects single-cell genomics to compositional data analysis, a mature statistical field with rigorous theoretical guarantees.
CytoSignal detects locations and dynamics of ligand–receptor signaling at cellular resolution from spatial transcriptomic data
Liu et al., Nature Genetics (2026). 10.1038/s41588-026-02624-9
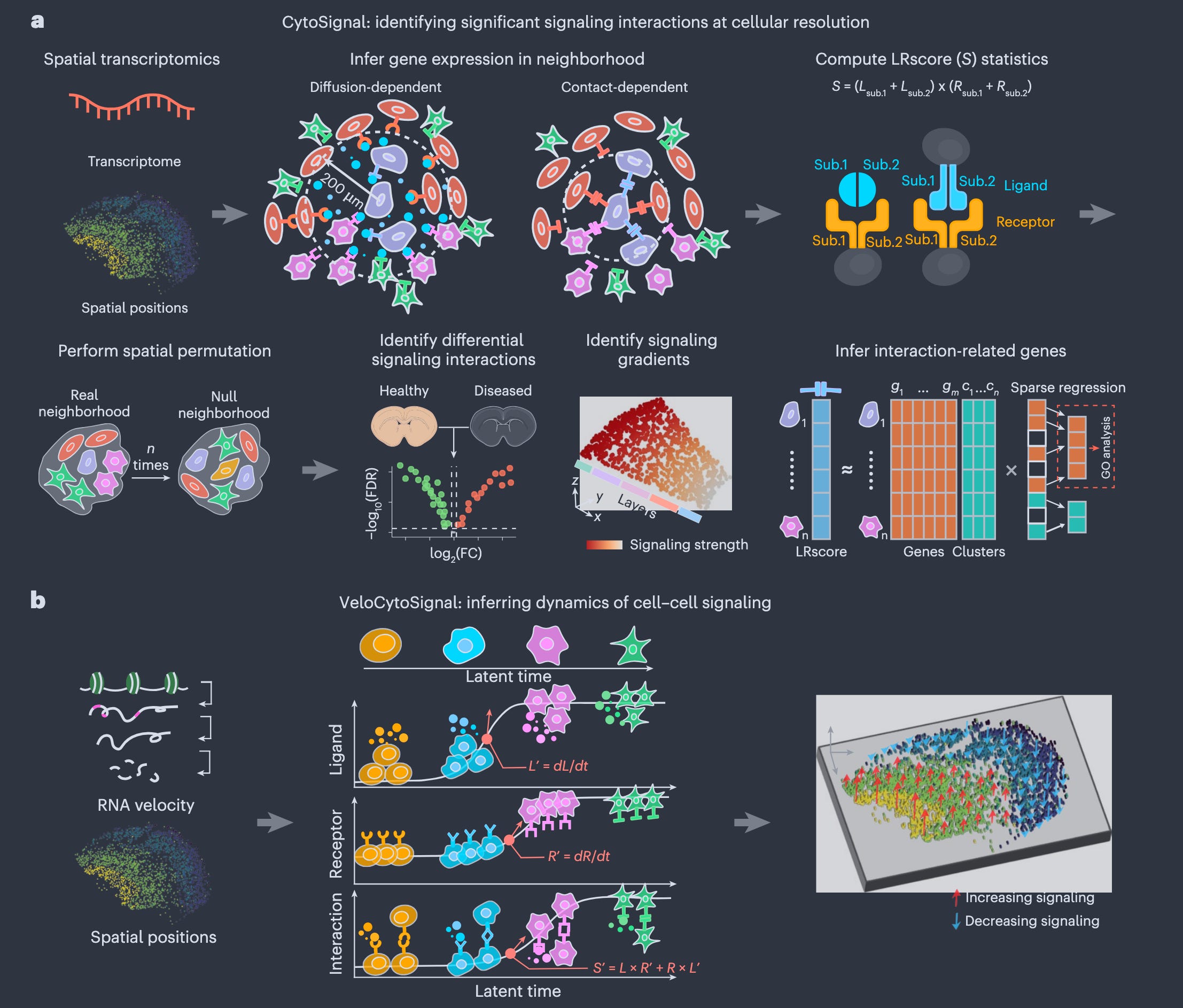
The paper in one sentence
CytoSignal is a computational framework that infers the locations, mechanisms, and temporal dynamics of ligand-receptor signaling interactions at single-cell resolution from spatial transcriptomic data, validated experimentally using proximity ligation assays.
Summary
This work introduces CytoSignal and VeloCytoSignal, two methods designed to analyze spatial transcriptomic datasets (e.g., Slide-seq, Stereo-seq, Visium HD) to detect and quantify cell-cell communication at cellular resolution. CytoSignal calculates a ligand-receptor (LR) signaling score (LRscore) for each spatial position, distinguishing between contact-dependent (requiring direct cell-cell contact) and diffusion-dependent (mediated by soluble ligands) interactions. It also identifies spatial gradients in signaling strength, signaling-associated genes, and differential signaling across conditions (e.g., age or disease). VeloCytoSignal extends this by predicting temporal dynamics of signaling activity using RNA velocity, enabling the inference of whether signaling is increasing or decreasing at each location. The authors validate their approach using proximity ligation assay (PLA), a gold-standard method for detecting protein-protein interactions in situ, demonstrating that CytoSignal’s predictions align more accurately with physical LR interactions than existing computational methods. They apply their tools to embryonic mouse brain and whole-embryo datasets, revealing biologically meaningful signaling patterns (e.g., Sema3a-PlexinA4 in neuronal migration, Dll1-Notch1 in choroid plexus development, and Fgf8-Fgfr1 in neural patterning). Additionally, they use CytoSignal to identify age-associated signaling changes in a mouse model of Parkinson’s disease, highlighting immune-related interactions like Spp1-Cd44.
Personal highlights
Cellular-resolution signaling inference: CytoSignal quantifies LR signaling activity at individual spatial positions, overcoming the limitations of previous methods that aggregate signals at the cluster or tissue level.
Mechanistic distinction: The method explicitly differentiates between contact-dependent (e.g., Efnb1-Epha4) and diffusion-dependent (e.g., Sema3a-PlexinA4-Nrp1) interactions, reflecting their distinct biological constraints.
Spatial gradient detection: CytoSignal identifies continuous gradients in signaling strength (e.g., Sema3a-PlexinA4 peaks in the subventricular zone of the mouse cortex), providing insights into how signaling varies across tissue layers.
Temporal dynamics with VeloCytoSignal: By integrating RNA velocity, VeloCytoSignal predicts whether signaling interactions are increasing or decreasing over time, validated using time-series Stereo-seq data (e.g., Alb-FcRn in liver and Wnt5a-Antxr1 in jaw/tooth development).
Experimental validation: PLA experiments confirm that CytoSignal’s predictions of LR interactions (e.g., Dll1-Notch1, Fgf8-Fgfr1) localize to the same tissue regions as physical protein-protein interactions, outperforming prior computational approaches in benchmarking tests.
Why should we care?
CytoSignal provides a scalable, statistically rigorous, and biologically interpretable way to map these interactions, distinguishing between signals that require direct contact (like a handshake) and those that can act over short distances (like a whispered message). This is not just a technical advance, it enables researchers to study how signaling drives development, disease, and tissue homeostasis in unprecedented detail. Critically, the authors validate their method experimentally using PLA, a technique that directly vizualizes protein interactions in tissue. This is a major step forward, as many computational methods in this field lack ground-truth validation. The finding that CytoSignal outperforms existing tools in predicting physical interactions suggests it could become a standard for spatial cell-cell communication analysis. The temporal component (VeloCytoSignal) adds another layer, revealing when these signals are ramping up or down, insights that could be vital for understanding dynamic processes like embryogenesis or tumour progression.

Other papers that peeked my interest and were added to the purgatory of my “to read” pile
scMTG reconstructs single-cell temporal dynamics with Markov transition generators
Cellpin enables reference-based imputation and denoising of spatial transcriptomes
Architectural fragility of gene regulatory networks underlies hematopoietic stem cell aging
Organoid-based colorectal tumor microenvironment model for immuno-oncology research
Spatial Gene Set Enrichment Analysis with Applications to Spatially Resolved Transcriptomic Data
Toward Interpretable and Generalizable AI in Regulatory Genomics
Evaluating agentic AI for biological discovery in autonomous and copilot settings
SPACE-seq integrates spatial transcriptomics and lineage tracing in native tissues
Molecular glue degraders of HuR suppress BRAF-mutant colorectal cancer
Back to basics: Observed statistics are sufficient to predict drug responses
Targeting Cancer-Specific Mutations with RNA-Triggered Chromatin Shredding
Whole-genome duplication shaped cell-type evolution in the vertebrate brain
Mitochondria directly interact with the nuclear pore complex
Biodiversity and biogeography of the multi-kingdom cancer microbiome
A conserved re-epithelialization program underlies malignancy in pancreatic ductal adenocarcinoma
Single-Cell Cross-Modal Transfer by Adversarial Fine-Tuning of Foundation Models
Querying Counterfactuals on Tissue Graphs with Supervised Disentanglement
The Challenge of Cell Segmentation in Spatially Resolved Transcriptomics
STITCH links cellular morphology and gene expression in spatial transcriptomics
Thanks for reading.
Cheers,
Seb.