

This week’s preprints and papers span from the molecular to the architectural—uncovering how cancer hijacks nerves and fibroblasts to resist therapy, how mechanical forces hardwire tumor plasticity, and how spatial and single-cell methods are evolving to make sense of tissues across scales. From novel biological insights (nerve injury as a driver of immunotherapy resistance, mitochondrial hand-offs that rewire fibroblasts) to cutting-edge computational tools (diffusion models for patient embeddings, graph optimization for spatial data), these studies highlight how cancer biology and computational innovation continue to reshape each other.
Preprints/articles that I managed to read this week
Cancer-induced nerve injury promotes resistance to anti-PD-1 therapy
Baruch et al. Nature (2025). https://doi.org/10.1038/s41586-025-09370-8
The paper in one sentence
Cancer cells damage surrounding nerves by degrading their protective myelin sheaths, which triggers a neuron-driven inflammatory response that creates an immunosuppressive environment and causes resistance to anti-PD-1 immunotherapy.
Summary
This comprehensive study reveals a novel mechanism of resistance to immune checkpoint blockade (anti-PD-1 therapy). The authors show that in multiple cancers (cutaneous squamous cell carcinoma, melanoma, gastric cancer), cancer cells directly injure tumor-associated nerves through a process called perineural invasion (PNI), specifically by degrading the myelin sheath. The injured neurons respond by autonomously initiating a healing response involving IL-6 and type I interferon (IFN-I) signaling. As the tumor grows, this nerve injury becomes chronic, and the local inflammation skews the entire tumor microenvironment into a suppressive and exhausted immune state, rendering anti-PD-1 therapy ineffective. Crucially, the study demonstrates that this resistance can be reversed by targeting this nerve-injury axis through surgical denervation, genetic knockout of key injury-response genes (Atf3, Ifnar1), or therapeutic blockade of IL-6 signaling.
Personal highlights
Cancer cells directly cause nerve injury via myelin degradation: using electron microscopy and electrophysiology, the authors provide striking visual and functional evidence that cancer cells strip away the protective myelin sheaths of nerves, leading to impaired nerve function—a process they term Cancer-Induced Nerve Injury (CINI).
Injured neurons are active immune orchestrators: the study shifts the paradigm of neurons as passive victims to active players in the tumor microenvironment. Injured neurons intrinsically upregulate and secrete key inflammatory cytokines like IL-6 and IFN-α, initiating an immunosuppressive cascade that attracts pro-healing M2 macrophages and exhausts T cells.
The immunosuppressive signal extends from the nerve niche throughout the tumor: through spatial transcriptomics and multi-optic mapping, they show that the immunosuppressive microenvironment created around injured nerves (the "perineural niche") correlates with and likely drives a similarly suppressive tone across the entire tumor.
Resistance is mechanistically reversible at multiple points: the causality of CINI in driving resistance is robustly proven by reversing the effect through surgical (denervation), genetic (Atf3 knockout in neurons, Ifnar1 global knockout), and pharmacological (anti-IL-6R antibody) interventions in vivo.
A conserved mechanism across neurotropic cancers:tThe association between a nerve injury gene signature and anti-PD-1 resistance is validated not only in their original cSCC cohort but also in independent, publicly available melanoma and gastric cancer datasets, suggesting a fundamental, tumor-agnostic biology
Why should we care?
This work fundamentally expands our understanding of the tumor microenvironment by identifying the nervous system as a key architect of immunotherapy resistance. It moves beyond the classic cancer-immune dialogue to a more complex "cancer-nerve-immune" triad. For oncologists and clinical researchers, it provides a compelling biological rationale for exploring novel combination therapies, such as adding IL-6 blockade to anti-PD-1, particularly for patients with cancers known for perineural invasion. It also offers potential biomarkers (e.g., nerve injury signatures) to predict who will respond to immunotherapy. For a broader audience, it highlights the remarkable interconnectedness of our bodily systems—showing how a process as specific as nerve healing can have devastating consequences by inadvertently helping cancer evade our immune system. This opens up an entirely new therapeutic avenue to overcome treatment resistance for many patients.
SAME: A New Method to Piece Together the Complex Puzzle of Multimodal Spatial Data
Pratapa et al. bioRxiv (2025). https://doi.org/10.1101/2025.07.12.664419
The paper in one sentence
SAME is a computational method that uses "space-tearing transforms" and integer linear programming to accurately align disparate spatial omics modalities (like protein, RNA, and metabolomics) from serial tissue sections, even when the tissue is torn, folded, or has changing anatomy.
Summary
Integrating data from different spatial omics technologies—each revealing a unique layer of molecular information (proteins, RNA, metabolites)—is a major challenge because the physical tissue sections are never perfectly identical. They can have tears, folds, and biological variations that break the topological assumptions of existing alignment tools. SAME (Spatial Alignment of Multimodal Expression) overcomes this by introducing a novel "space-tearing transform" framework. Instead of forcing a perfect, smooth alignment, SAME uses an integer linear programming (ILP) approach to maximize matches between cell types across modalities while selectively allowing local topological disruptions where the data justifies it. It monitors these disruptions through the "flipping" of triangles in a spatial mesh. This enables robust, tunable integration that preserves biological meaning, unlocking the discovery of cryptic cell states and spatial niches that are invisible to any single modality.
Personal highlights
Topology-flexible alignment via integer linear programming: SAME formulates the alignment problem as an ILP that directly optimizes for cell-type correspondence while penalizing unnecessary geometric distortions. This provides a principled, tunable framework for spatial integration that is both rigorous and flexible.
Introduction of "space-tearing transforms": the key innovation is a class of transforms that allows for controlled, localized topological violations (like "tears" or "folds" in the tissue map) within an otherwise coherent alignment, a critical capability for handling real-world tissue artifacts that break existing methods.
Geometric proxy for topological violation: SAME cleverly uses the signed area of triangles in a Delaunay triangulation to detect when a local spatial relationship has been inverted. This provides a simple yet powerful geometric measure to monitor and control for space-tearing events during optimization.
Modality-agnostic phenotype matching: instead of trying to align raw, often incompatible molecular counts (e.g., RNA vs. protein levels for the same gene), SAME matches cells based on their inferred phenotypes (cell types). This approach seamlessly integrates diverse data types, from probabilistic RNA-based classifications to definitive protein marker calls and even morphology-based features from H&E images.
Unlocks multimodal discovery in complex tissues: by providing robust alignment, SAME enables the fusion of complementary data strengths. The authors demonstrate this by revealing niche-specific T cell states in tongue and lung cancer by integrating protein (for confident identity) with RNA (for functional state), and by uncovering localized metabolic crosstalk between tumor cells and macrophages by integrating proteomics with metabolomics.
Why should we care?
SAME tackles a fundamental bottleneck in the rapidly growing field of spatial biology: how to confidently combine the pieces of the molecular puzzle gathered from different experiments. For researchers, it provides a much-needed, robust tool that respects the messy reality of tissue samples, allowing them to design better experiments where each modality can be processed under optimal conditions. By enabling true multimodal integration, SAME moves beyond simply cataloging what molecules are where and begins to reveal the functional relationships between them—like how a specific immune cell's location in a tumor dictates its behavior or how cancer cells metabolically communicate with their neighbors.
scSet: Bridging the Single-Cell to Patient Gap with Diffusion Models
Boiarsky et al. bioRxiv (2025). https://doi.org/10.1101/2025.08.21.671613
The paper in one sentence
scSet is a diffusion-based autoencoder that learns robust, clinically informative patient-level representations from the unordered set of single cells in a sample by using a transformer to encode and a conditional diffusion model to decode, enabling superior performance on downstream prediction tasks even with limited labeled data.
Summary
Translating single-cell RNA sequencing (scRNA-seq) data into patient-level clinical insights is challenging because a patient's data is an unordered, variable-sized set of cells, and labeled datasets are small. scSet overcomes this by framing the problem as an autoencoding task for sets. It uses a transformer-based encoder to process any number of input cells and produce a fixed-size patient embedding. The key innovation is using a conditional Denoising Diffusion Probabilistic Model (DDPM) as the decoder. This diffusion model learns to generate a realistic set of cells that match the patient's biological profile from noise, conditioned on the patient embedding. This self-supervised "reconstruction" task, trained on large, unlabeled datasets, forces the model to learn meaningful patient representations that capture cellular composition and state. These representations can then be efficiently fine-tuned with a simple classifier on small, labeled datasets to predict clinical outcomes, significantly outperforming existing methods that rely on simple averaging or supervised-only approaches.
Personal highlights
Diffusion-based decoding for set-structured data: the core methodological innovation is using a conditional diffusion model as the decoder in an autoencoder framework. This elegantly handles the unordered, variable-sized nature of single-cell data by learning to generate a realistic set of cells that is representative of the input patient sample.
Transformer-based permutation-invariant encoding: employs a transformer encoder to process the entire set of cells from a patient. The self-attention mechanism captures complex, higher-order interactions between cells, and the use of a [CLS] token provides a natural, permutation-invariant summary to produce the final patient embedding.
Self-supervised pre-training on unlabeled data: leverages a noise prediction loss to train the entire autoencoder (encoder + diffusion decoder) on large-scale, unlabeled scRNA-seq datasets. This pre-training step is crucial for learning robust, general-purpose patient representations that capture biologically meaningful variation without the need for costly clinical labels.
Effective knowledge transfer to data-scarce clinical tasks: demonstrates that the pre-trained patient embeddings serve as powerful features for downstream prediction. When fine-tuned on small labeled cohorts (e.g., 25-100 patients), they consistently outperform supervised baselines, highlighting the value of pre-training for overcoming the label-scarcity problem in computational pathology.
Ablations validating the architectural choices: includes critical controlled experiments showing that both the transformer architecture and the diffusion-based pre-training objective contribute significantly to the final performance, outperforming alternative encoders (like ABMIL) and decoders (like flow-based models).
Why should we care?
scSet addresses a fundamental bottleneck in modern computational biology: how to move from rich, single-cell observations to actionable, patient-level insights. For researchers and clinicians, it provides a powerful tool that maximizes the value of expensive scRNA-seq data. By learning from vast amounts of unlabeled data, it reduces the need for large, labeled clinical cohorts, which are difficult and expensive to assemble. The resulting patient embeddings capture subtle but clinically relevant biological signals, like shifts in cellular composition or state, that are often lost when data is simplistically averaged. This enables more accurate prediction of disease states, treatment responses, and patient outcomes from complex cellular data. For the field, scSet represents a significant step towards a foundational model for single-cell data, providing a flexible, pre-trained backbone that can be adapted to a wide range of translational research questions.
Tracing colorectal malignancy transformation from cell to tissue scale
Crowell et al. bioRxiv (2025). doi: 10.1101/2025.06.23.660674
The paper in one sentence
This study charts how normal colon tissue evolves into colorectal cancer by combining whole-transcriptome spatial transcriptomics, single-nucleus RNA-seq, and digital pathology to reveal the cellular, molecular, and architectural dynamics of tumor progression and spread.
Summary
Crowell et al. present one of the most detailed multi-scale maps of colorectal cancer (CRC) initiation and progression to date. Using CosMx whole-transcriptome spatial molecular imaging, snPATHO-seq, and histopathology, they profiled ~3.5 million cells across normal mucosa, adenomas, carcinomas, and a matched lymph node metastasis. They identified 43 epithelial, immune, and stromal subpopulations and reconstructed the malignant trajectory from healthy crypts to invasive tumors. Key findings include the bifurcation into LGR5+ stem-like tumor cores versus MMP7+ fetal-like invasive fronts, the definition of “transition crypts” as single crypts undergoing abrupt transformation, and the tracing of fetal-like tumor cells with fibroblast and macrophage partners to vascular invasion sites and lymph node metastases. The work highlights how cellular programs, spatial architecture, and microenvironmental interactions co-evolve during colorectal tumorigenesis.
Personal highlights
Bifurcating tumor evolution into core and invasive fronts: CRC development splits into two trajectories—dense, LGR5+ stem-like tumor cores versus MMP7+ fetal-like invasive margins enriched with cancer-associated fibroblasts and immunosuppressive macrophages.
Transition crypts as single-crypt transformation events: identifies “transition crypts” where individual colonic crypts display mixed normal and malignant features, marking abrupt local shifts in cellular identity and tumor progression.
Spatial niches organizing malignancy: reveals concentric layers of epithelial, stromal, and immune subpopulations that create tumor cores, invasive fronts, and immunosuppressive microenvironments aligned with histological grading.
Linking invasive programs to metastasis: tracks fetal-like MMP7+ tumor cells with FAP+ fibroblasts from primary tumor invasive fronts into blood vessels and matched lymph node metastases, directly connecting local invasion to dissemination.
Orthogonal resolution of pathology: shows how spatial transcriptomics provides molecular clarity in regions where histopathology alone was ambiguous, integrating tissue architecture with transcriptional dynamics.
Why should we care?
This work demonstrates how spatially resolved, single-cell approaches can bridge the gap between traditional histopathology and molecular biology in cancer research. For pathologists, it refines tumor grading by providing transcriptional confirmation of malignant states; for biologists, it uncovers how stem-like and invasive tumor programs interact with the microenvironment to drive progression and metastasis. Clinically, it highlights early premalignant states (such as EMP1+ cells) with potential links to relapse, and underscores the role of tumor–stroma–immune crosstalk in invasion. More broadly, it exemplifies how multi-scale, spatially anchored maps can transform our understanding of cancer evolution, from single crypts to tissue-wide architecture, and may inform future diagnostics, prognosis, and therapeutic strategies.
MIRO2-mediated mitochondrial transfer from cancer cells induces cancer-associated fibroblast differentiation
Cangkrama et al. Nat Cancer (2025). https://doi.org/10.1038/s43018-025-01038-6
The paper in one sentence
Cancer cells hijack a protein called MIRO2 to send their mitochondria to nearby fibroblasts, reprogramming them into tumor-promoting allies that fuel cancer growth.
Summary
This study uncovers a new way cancer cells manipulate their surroundings to support tumor growth. Researchers found that various cancer cells can physically transfer their energy-producing mitochondria to neighboring fibroblasts (a common cell type in connective tissue). This transfer happens through tiny tunnels called tunneling nanotubes (TNTs). Once the fibroblasts receive these cancer-derived mitochondria, they undergo a dramatic transformation, becoming "cancer-associated fibroblasts" (CAFs), a cell type known to support tumors by promoting inflammation, remodeling tissue, and secreting growth factors. The study identifies the mitochondrial trafficking protein MIRO2 as the essential molecular engine behind this transfer. Cancer cells at the invasive edge of tumors overexpress MIRO2, and blocking it prevents mitochondrial donation, CAF formation, and tumor growth in mouse models. This work reveals mitochondrial transfer as a direct cell-to-cell communication strategy that tumors use to build a supportive microenvironment.
Personal highlights
Direct mitochondrial donation as a driver of stromal activation: the study moves beyond secretome-mediated signaling to show that the physical transfer of entire organelles—functional mitochondria—from cancer to stromal cells is a potent direct mechanism for inducing a pro-tumorigenic fibroblast state.
MIRO2 as the master regulator of intercellular mitochondrial trafficking: identifies the Rho GTPase MIRO2, not its homolog MIRO1, as the critical and specific driver of mitochondrial transfer from cancer cells to fibroblasts, positioning it as a compelling therapeutic target.
Functional reprogramming beyond marker expression: the transferred mitochondria do more than just change fibroblast gene signatures; they functionally rewire the recipient cell's metabolism, boosting oxidative phosphorylation and ATP production, which in turn fuels proliferation and the secretion of tumor-promoting factors.
A threshold effect in cellular reprogramming: induction of the CAF phenotype exhibits a saturable, dose-dependent response to mitochondrial uptake, suggesting a biological threshold that must be crossed for reprogramming to occur, beyond which additional mitochondria have diminishing returns.
Clinical relevance from single-cell to spatial transcriptomics: leveraging human patient data, the study shows that MIRO2 is specifically overexpressed in invasive tumor cells at the leading edge of cancers, spatially correlating with CAF-rich regions, which underscores the clinical significance of this pathway.
Why should we care?
This work fundamentally changes how we think about the conversation between a tumor and its surrounding tissue. It shows that cancer cells don't just send chemical signals; they can directly donate parts of their cellular machinery to reprogram their neighbors into loyal supporters. The identification of MIRO2 as the key enabler offers a new and precise therapeutic strategy: by blocking this protein, we could potentially cut off this line of communication, preventing tumors from building their supportive ecosystem and slowing their growth. For cancer biologists, it reveals a novel mechanism of tumor-stroma crosstalk; for clinicians, it highlights MIRO2 as a promising new target, especially for stroma-rich cancers like pancreatic cancer where current treatments often fail.
Mechanical confinement governs phenotypic plasticity in melanoma
Hunter et al. Nature (2025). https://doi.org/10.1038/s41586-025-09445-6
The paper in one sentence
This study reveals that physical squeezing by the tumor microenvironment triggers melanoma cells to switch from a proliferative state to an invasive, drug-resistant "neuronal" state by activating the DNA-bending protein HMGB2, which remodels chromatin to promote this dangerous transformation.
Summary
This research uncovers a fundamental mechanism of cancer cell plasticity. Using zebrafish models and human patient samples, the authors identify a subpopulation of "interface" melanoma cells at the tumor's edge that are physically confined by surrounding tissues. This mechanical pressure causes the cells' nuclei to elongate and triggers a dramatic reprogramming. The cells disassemble their pigment-producing machinery, exit the cell cycle, and activate a neuronal gene program, complete with a protective cage of acetylated tubulin around the nucleus. The key mediator of this switch is the protein HMGB2, which is upregulated by the force and stabilizes on chromatin to increase accessibility at pro-invasive and neuronal genes. Functionally, this HMGB2-high state makes cells more invasive but less proliferative and, crucially, more resistant to targeted therapy. The work provides a direct link between the physical microenvironment and epigenetic-driven phenotypic plasticity in cancer.
Personal highlights
Mechanical force as a master regulator of cell state: the study elegantly demonstrates that simple physical confinement—mimicking the pressure at a tumor's edge—is sufficient to trigger a stable, invasive phenotypic switch, moving beyond soluble chemical signals as the primary drivers of plasticity.
Cancer cells hijack a neuronal survival program: confined melanoma cells assemble a protective perinuclear "cage" of stabilized, acetylated tubulin, a mechanism co-opted from migrating neurons that must also squeeze through tight spaces, highlighting a fascinating evolutionary convergence.
Chromatin as a mechanical sensor: the DNA-bending protein HMGB2 is identified as the critical link. Force prolongs its contact with chromatin, and this prolonged contact itself alters the chromatin landscape to favor the expression of invasive genes, positioning the nucleus as a central mechanosensory organelle.
The trade-off is physically enforced: the work provides a mechanistic basis for the classic "go or grow" hypothesis. Confinement, via HMGB2, actively suppresses proliferation pathways while simultaneously enhancing invasion, explaining the mutual exclusivity of these states at the single-cell level.
Direct therapeutic implications: the confined, HMGB2-high state confers resistance to both chemotherapy (Taxol) and targeted therapy (dabrafenib/trametinib), identifying mechanical confinement not just as a driver of spread but also as a key contributor to treatment failure.
Why should we care?
This work fundamentally changes how we think about cancer progression. It argues that the physical "crush" of a growing tumor isn't just a passive barrier; it's an active instructor that tells cells to become aggressive and treatment-resistant. For patients, this suggests that a tumor's physical structure could be a predictor of its behavior and a new avenue for therapy, could we make the environment less restrictive or block the cell's ability to sense squeezing? For scientists, it beautifully bridges biophysics and genetics, showing how mechanical forces are translated into lasting epigenetic changes that dictate cell fate. It’s a compelling reminder that to understand cancer, we need to consider not just the genetic code within the cell, but also the physical world that surrounds it.
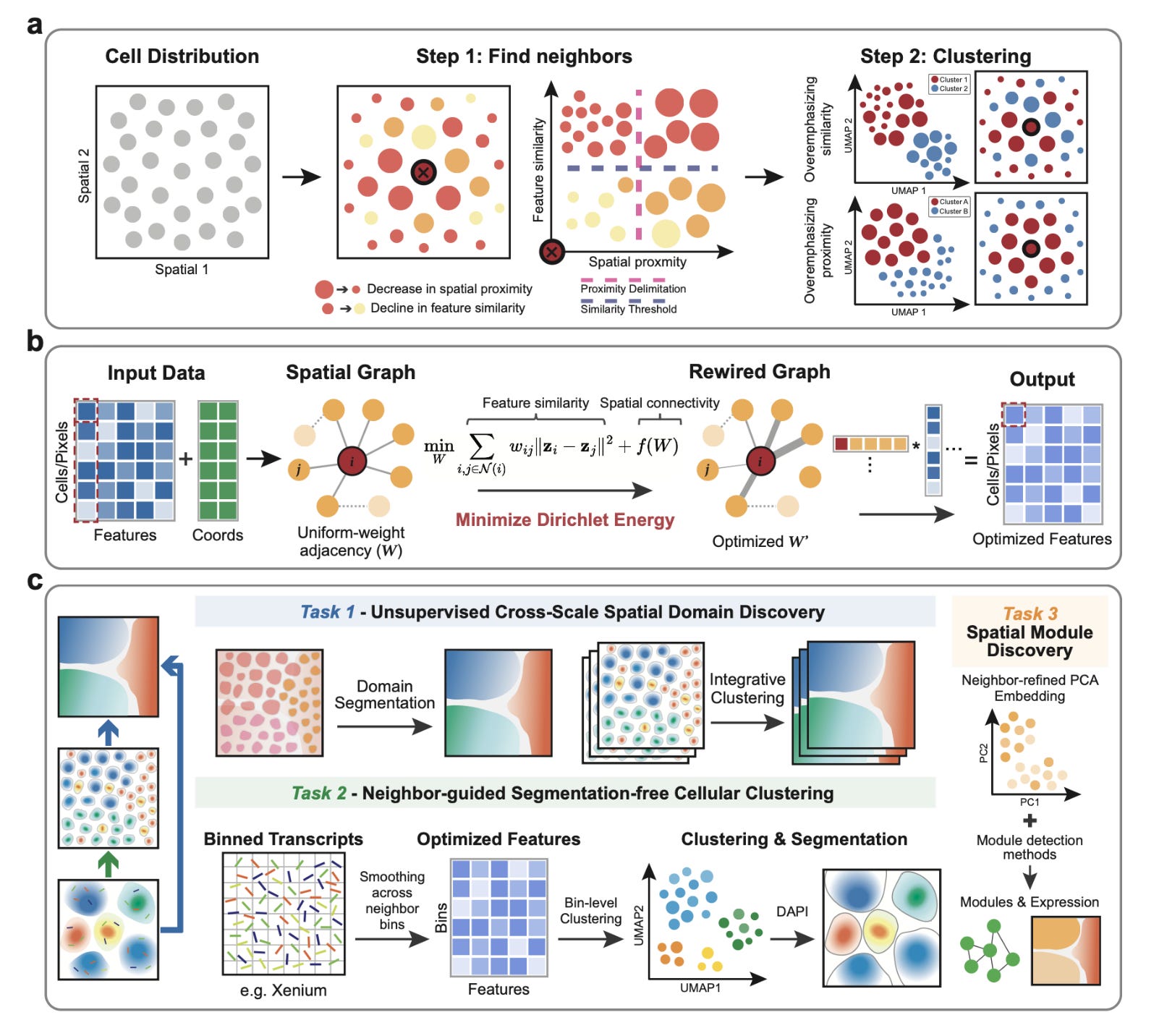
Energy-Regularized Graph Learning for Multiscale Spatial Representation
Gong et al. bioRxiv (2025). https://doi.org/10.1101/2025.08.22.671846
The paper in one sentence
Glimmer is a unified framework that learns adaptive neighborhood graphs by minimizing Dirichlet energy to create spatially-informed embeddings, enabling everything from tissue-scale domain discovery to segmentation-free, subcellular transcript assignment across diverse spatial omics technologies.
Summary
Spatial omics technologies generate complex, high-dimensional data where a cell's molecular identity is intrinsically linked to its physical location. Existing methods struggle to balance these two signals: over-emphasizing molecular similarity creates "salt-and-pepper" clusters, while over-smoothing based on location can blur important biological differences. Glimmer solves this by reframing the problem as a graph optimization task. Starting from a simple spatial neighbor graph, it uses a principled Dirichlet energy minimization approach with a log-barrier regularizer to iteratively reweight the connections between cells. This process automatically strengthens edges between cells that are both spatially close and molecularly similar, while weakening others. The result is a context-aware, adaptive graph that provides a denoised, spatially coherent embedding of the data. This single framework is remarkably versatile, outperforming other methods at identifying tissue domains in Slide-seq and MERFISH data, revealing niche-specific immune states in melanoma, and enabling highly accurate, segmentation-free clustering of transcripts at subcellular resolution in Xenium data, correcting common cell segmentation errors.
Personal highlights
Unified graph optimization across scales and modalities: Glimmer's core innovation is its elegant reformulation of spatial representation learning as a Dirichlet energy minimization problem. This single, principled mathematical framework is applied consistently from millimeter-scale tissue regions down to micron-scale transcript bins, making it universally applicable across technologies like Slide-seq, MERFISH, Xenium, and CODEX without platform-specific tweaks.
Adaptive, data-driven neighbor weighting: instead of using a fixed kernel or heuristic, Glimmer learns the optimal weight for every single edge in the neighborhood graph. This allows it to dynamically balance molecular similarity and spatial proximity in a context-aware manner, preventing over-smoothing in heterogeneous regions (like tumor boundaries) while still enforcing coherence in homogeneous zones (like brain layers).
Segmentation-free cellular deconvolution at subcellular resolution: Glimmer directly addresses a major pain point in imaging-based spatial transcriptomics (e.g., Xenium) by bypassing error-prone cell segmentation altogether. It aggregates transcripts into fine bins, applies its adaptive smoothing, and clusters the bins to reassign transcripts with high accuracy, significantly improving cell type-specific marker expression and purity over existing segmentation-based and segmentation-free methods.
Explicit tunability from expression-driven to niche-driven regimes: the log-barrier regularizer provides a clear, interpretable knob to tune the embeddings. Researchers can smoothly transition from a primarily expression-driven view (useful for identifying cell states) to a niche-driven view (useful for identifying anatomical domains), with a balanced regime in between that reveals spatially-informed biological structures, like distinct exhausted and cytotoxic T cell niches in a tumor.
Revealing niche-specific biology and communication: the power of Glimmer's embeddings is demonstrated by its ability to uncover biologically meaningful insights. It not only identifies fine-grained anatomical zones in human tonsils (like dark and light zones of germinal centers) but also enables the subsequent analysis of zone-specific gene expression and cell-cell communication networks, linking spatial architecture directly to function.
Why should we care?
Glimmer tackles a fundamental and pervasive challenge in spatial biology: how to faithfully represent data where a cell's location and its molecular state are two sides of the same coin. For bioinformaticians, it provides a robust, scalable, and interpretable framework that outperforms existing GNN and kernel-based methods, setting a new standard for spatial representation learning. For experimentalists, its ability to generate accurate, segmentation-free cell maps from Xenium data is a direct solution to a major technical hurdle, ensuring that downstream analyses are built on a solid foundation. Most importantly, by providing a clearer, denoised view of tissue organization across scales, Glimmer empowers researchers to ask more precise questions about how cellular ecosystems are organized in health and disease, ultimately leading to better mechanistic insights into development, immunology, and oncology.

Other papers that peeked my interest and were added to the purgatory of my “to read” pile
FSP1 and histone deacetylases suppress cancer persister cell ferroptosis
Uncovering Developmental Lineages from Single-cell Data with Contrastive Poincaré Maps
Rigorous integration of single-cell ATAC-seq data using regularized barycentric mapping
rbio1 - training scientific reasoning LLMs with biological world models as soft verifiers
A Benchmark of Semi-Supervised scRNA-seq Integration Methods in Real-World Scenarios
Thanks for reading.
Cheers,
Seb.