
Weekly reads 15/06/26
From spatial counterfactuals to tumor microbiomes: how context reshapes biology

This week’s reads focused on papers that push the boundaries of spatial and single-cell biology, revealing how context, whether spatial, genetic, or microbial, shapes cellular behavior and disease. Cellina unveils a novel method of tissue graph counterfactuals by means of supervised disentanglement to guess the cellular behavior upon change in microenvironment, while Decima decodes the sequence determinants of gene expression across 22 million single-cell profiles, linking DNA to disease-specific regulation. With respect to cancer, a spatial atlas of muscle-invasive bladder cancer reveals a luminal-to-basal differentiation axis which controls the immune architecture and unveils therapeutic vulnerabilities, and DeLeakage after spatial transcriptomics discovers a widespread transcript leakage issue, therefore restores the accurate interpretation of the results. At the same time, Cheruiyot et al. present how inflammatory cytokines such as IFNγ render tumors dependent on previously unknown factors, and Dohlman et al. confirm what many have questioned over the years: only orodigestive cancers contain microbiomes that are detectable, site-specific, and that have significant implications for genomic instability
Preprints/articles that I managed to read this week
Querying Counterfactuals on Tissue Graphs with Supervised Disentanglement
Moeed et al. arxiv (2026). https://arxiv.org/abs/2606.08493
The paper in one sentence
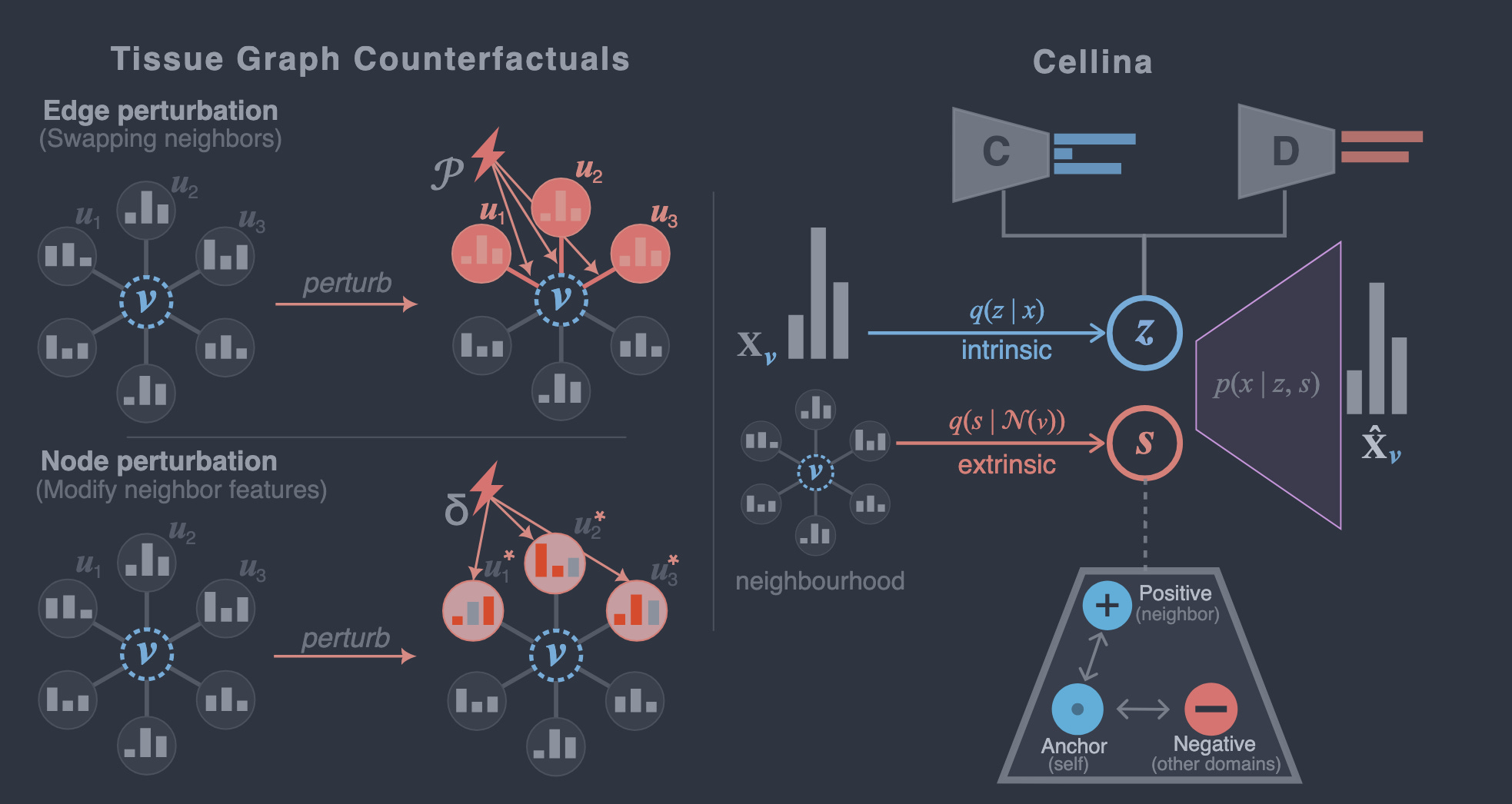
This work introduces Cellina, a supervised disentanglement framework that formalizes tissue graph counterfactuals as spatial interventions (edge and node perturbations) and enables accurate prediction of how cells would respond to altered neighborhood contexts in tissues.
Summary
Spatial transcriptomics has revealed that a cell’s gene expression is strongly influenced by its local microenvironment, yet most computational models still treat cells as independent entities. This paper addresses this gap by formalizing tissue graph counterfactuals, a class of spatial interventions that either rewire connections between cells (edge perturbations) or modify the expression of neighboring cells (node perturbations). The authors propose Cellina, a graph variational autoencoder (VAE) that decomposes each cell’s gene expression into two latent components: an intrinsic representation (encoding cell identity) and an extrinsic (spatial) representation (encoding microenvironmental influence). Supervised disentanglement, guided by cell-type and spatial-domain labels, ensures that these components remain biologically interpretable. Across benchmarks spanning 2.5 million spatially resolved cells from colorectal cancer and mouse brain datasets, Cellina outperforms both spatially informed and non-spatial baselines in counterfactual prediction, disentanglement quality, and scalability. Additionally, Cellina identifies biologically distinct cancer subdomains and enables targeted pathway-specific neighbor perturbations, demonstrating its utility for both discovery and simulation.
Personal highlights
Formalization of tissue graph counterfactuals: The paper provides a unified definition of spatial interventions as either edge perturbations (rewiring neighborhood topology) or node perturbations (altering neighbor gene expression), establishing a common framework for studying neighborhood-driven cell responses.
Supervised disentanglement: Cellina uses a dual-encoder graph VAE with biological supervision (cell-type and spatial-domain labels) to explicitly separate intrinsic cell identity from extrinsic spatial context, improving interpretability and predictive performance.
Scalable and robust performance: Across large-scale datasets, Cellina consistently outperforms competitors on key metrics (e.g., Pearson r, Signed Precision, RMSE) for both edge and node perturbation tasks, demonstrating generalizability across tissues and species.
Biological discovery without supervision: The spatial latent representation in Cellina reveals distinct cancer subdomains with interpretable signaling programs (e.g., TGFβ-dominant vs. NFκB/MAPK-dominant), aligning with known biological mechanisms and published findings.
Pathway-targeted in silico perturbations: Cellina enables simulations of pathway-specific neighbor perturbations, recreating observed subdomain effects (e.g., up-regulation of FN1 and MMP3 in cancer-associated fibroblasts) and linking them to established biological pathways.
Why should we care?
This work pushes the boundaries of computational spatial biology by providing a framework to simulate how cells would behave in altered microenvironments, disease progression, and therapeutic interventions. Traditional single-cell models often ignore the spatial context that shapes cellular behavior, but Cellina explicitly accounts for it, offering a more realistic representation of biological systems. However, the approach has limitations: it relies on computational simulations rather than direct experimental validation, and its effectiveness depends on the quality of spatial annotations and assumptions about cell segmentation and neighborhood definitions.
Decoding sequence determinants of gene expression in diverse cellular and disease states
Lal et al. Nature Methods (2026). 10.1038/s41592-026-03102-0
The paper in one sentence
Decima, a deep learning model trained on over 22 million single-cell RNA-seq profiles, predicts cell type- and disease-specific gene expression from DNA sequence, enabling the interpretation of regulatory mechanisms and the effects of noncoding variants at unprecedented resolution.
Summary
This study introduces Decima, a sequence-to-function model that leverages single-cell and single-nucleus RNA-seq data from 22 million cells across 201 cell types, 271 tissues, and 82 diseases. Decima predicts gene expression from 524-kb DNA sequences surrounding gene transcription start sites, achieving high accuracy (mean Pearson correlation of 0.80 for held-out genes across pseudobulks). The model identifies cell type-specific regulatory elements, transcription factor motifs, and sequence determinants of disease-associated expression changes. It also predicts the impact of noncoding variants (e.g., eQTLs and GWAS hits) at cell type resolution, outperforming bulk-trained models like Borzoi. Finally, Decima demonstrates proof-of-concept potential for designing synthetic regulatory elements with cell type- and disease-specific activity.
Personal highlights
Scalable single-cell resolution: Decima is trained on pseudobulk aggregated sc/snRNA-seq data from 22M cells, enabling predictions across 201 cell types, 271 tissues, and 82 diseases, exceeding the scope of previous bulk-trained models.
Accurate cell type-specific predictions: The model achieves a mean Pearson correlation of 0.80 for predicting expression of held-out genes across pseudobulks and 0.58 for predicting expression of the same gene across different cell types.
Interpretability of regulatory mechanisms: Using input × gradient attribution, Decima highlights cell type-specific regulatory elements (e.g., promoters, enhancers) and transcription factor motifs (e.g., C/EBP, RXR, TWIST1) driving differential expression, even for distal elements (>100 kb from TSS).
Variant effect prediction at cell type resolution: Decima prioritizes cell type-specific eQTLs and GWAS variants, correctly predicting the direction of effect for 87% of high-confidence sc-eQTLs and linking variants to relevant cell types (e.g., immune cells for autoimmune diseases, hepatocytes for triglyceride-associated variants).
Disease and design applications: The model reveals sequence determinants of disease-specific cell states (e.g., inflammatory signatures in Crohn’s disease fibroblasts) and demonstrates potential for in silico design of synthetic regulatory elements with cell type- and disease-biased activity.
Why should we care?
This work is connecting DNA sequence to functional outcomes in specific cell types and disease contexts. While bulk models like Borzoi have advanced our understanding of gene regulation, they lack the resolution to dissect mechanisms in heterogeneous tissues or pathological states. Decima’s ability to predict expression from sequence at single-cell resolution enables: precision medicine, disease mechanism discovery and synthetic biology. While Decima is a major step forward, its reliance on pseudobulk aggregation means it may miss nuanced single-cell heterogeneity. The model’s performance also depends on the quality and diversity of training data, and its predictions are limited to the cell types, tissues, and diseases represented in the atlas. Additionally, distal enhancer-gene interactions remain challenging, and experimental validation is still required for applications like regulatory element design
A Spatial Atlas of Muscle-Invasive Bladder Cancer Reveals Lineage-Specific Vulnerabilities and Immune Architecture
Yu et al., Cancer Discovery (2026). DOI: 10.1158/2159-8290.CD-26-0099
The paper in one sentence
This study constructs a spatially resolved atlas of muscle-invasive bladder cancer (MIBC), uncovering a continuous luminal-to-basal differentiation axis that shapes tumor architecture, immune organization, and lineage-specific therapeutic vulnerabilities.
Summary
Using spatial transcriptomics on 22 pre-treatment MIBC tumors—integrated with matched bulk RNA-seq, whole-exome sequencing, and single-cell RNA-seq—the authors map a spatially organized luminal-to-basal axis within individual tumors. Luminal tumor cores, enriched for FGFR3 and NECTIN4, are immune-excluded and associated with proteotoxic stress responses, while basal-like states at invasive margins exhibit EGFR signaling, epithelial-mesenchymal transition (EMT), genomic instability, and dense immune infiltration (including tertiary lymphoid structures, TLSs). The study validates these findings across >3,000 tumors, demonstrating conserved FGFR3-EGFR lineage exclusivity and associating luminal states with vulnerability to NECTIN4-targeted therapies (e.g., enfortumab vedotin) and basal states with chemotherapy sensitivity. Functional experiments further show that FGFR3 and EGFR signaling reciprocally regulate lineage identity, and that TLS maturity and proximity to basal regions correlate with response to neoadjuvant chemotherapy.
Personal highlights
Spatially organized lineage plasticity: MIBC tumors harbor a continuous luminal-to-basal differentiation axis, with luminal states concentrated in tumor cores and basal-like states at invasive margins, revealing intratumoral heterogeneity that bulk profiling cannot capture.
Opposing RTK programs define lineage identity: FGFR3 and EGFR mark opposing poles of the differentiation spectrum, with mutual exclusivity across tumors and conserved associations with luminal and basal programs, respectively.
Immune architecture linked to lineage states: Basal-like regions co-localize with immune-rich, immunosuppressive niches (including TLSs), whereas luminal regions are immune-excluded, providing a spatial explanation for divergent responses to immunotherapy and chemotherapy.
NECTIN4 as a luminal-specific target: NECTIN4 is spatially restricted to luminal cores, predicting sensitivity to antibody-drug conjugates like enfortumab vedotin, and its overexpression induces a luminal-like, immune-quiescent phenotype in vitro.
TLS heterogeneity and therapeutic relevance: Peritumoral TLSs in chemotherapy responders exhibit immune-active states (e.g., interferon signaling), while those in non-responders show immunosuppressive features (e.g., Treg exhaustion markers), suggesting TLS maturation as a biomarker for treatment response.
Why should we care?
This work challenges the oversimplified view of bladder cancer as a binary disease (luminal vs. basal) by demonstrating that individual tumors contain a structured, radial gradient of cell states, with distinct biological and clinical implications. The spatial separation of luminal (differentiated, immune-cold) and basal (plastic, immune-hot) regions explains why patients with the same bulk subtype may respond differently to therapies: luminal cores may evade immune-based treatments but remain vulnerable to NECTIN4-targeted drugs, while basal margins, rich in immune cells, may be more susceptible to chemotherapy or immunotherapy. Critically, the study also highlights limitations of static spatial profiling: the snapshot nature of the data cannot capture temporal dynamics (e.g., how lineages evolve during therapy), and the 2D sections may not fully represent 3D tumor architecture. Nonetheless, the identification of FGFR3/EGFR as lineage gatekeepers and TLS maturity as a response predictor offers actionable insights for spatially informed precision oncology.
Inflammatory cytokines induce new cancer dependencies
Cheruiyot et al., Nature Genetics (2026). 10.1038/s41588-026-02614-x
The paper in one sentence
Inflammatory cytokines like interferon-γ (IFNγ) and interferon-β (IFNβ) induce tumor-intrinsic genetic vulnerabilities, revealing the GPI transamidase complex and the lipid phosphatase FITM2 as critical dependencies that sensitize cancer cells to immune checkpoint blockade (ICB) and cytokine-mediated stress.
Summary
This study uses genome-scale CRISPR loss-of-function screens in eight syngeneic mouse cancer models (melanoma, pancreatic, renal, lung, and colorectal) to map genetic dependencies induced by inflammatory cytokines (IFNβ, IFNγ, TNF). The authors identify context-specific vulnerabilities: the GPI transamidase complex (Gpaa1, Pigk, Pigu, Pigt, Pigs) as a dependency for resistance to type I/II IFNs, and FITM2—a regulator of ER lipid homeostasis, as a selective dependency for IFNγ. Loss of these genes sensitizes tumors to cytokines in vitro and enhances responses to ICB in vivo. Mechanistically, FITM2 deficiency triggers ER and oxidative stress in response to IFNγ, leading to a paraptosis-like cell death mediated by interferon-inducible GTPases (IRGs and GBPs). The GPI transamidase complex, on the other hand, restrains IFN sensitivity via BST2/tetherin, a viral restriction factor. While the study provides a robust preclinical framework, clinical validation in human ICB-treated cohorts remains limited due to low mutation frequencies and lack of statistical power.
Personal highlights
Cytokine-specific dependencies mapped at scale: Genome-wide CRISPR screens across eight mouse tumor models exposed to IFNβ, IFNγ, or TNF reveal shared and model-specific genetic vulnerabilities, including canonical IFN signaling components (e.g., Socs1, Ptpn2, Usp18) and novel targets like Gpaa1 and Fitm2.
GPI transamidase complex as an IFN resistance mechanism: The GPI transamidase complex (required for GPI-anchor protein biosynthesis) suppresses tumor sensitivity to both type I (IFNβ) and type II (IFNγ) interferons via BST2/tetherin, a previously unrecognized link between IFN sensing and GPI-anchored proteins.
FITM2 loss drives IFNγ-induced ER and oxidative stress: FITM2-deficient tumors accumulate ER stress (UPR activation, BiP upregulation) and mitochondrial oxidative stress (glutathione metabolism, SOD1 dependency), culminating in a paraptosis-like cell death characterized by cytoplasmic vacuolization and caspase-independent lysis.
Interferon-inducible GTPases (IIGTPases) as executioners: IFNγ induces IRGs (Irgm1/Irgm2) and GBPs (Gbp6/7/8) in FITM2-null cells, which are essential for triggering ER stress and oxidative damage, revealing a host-defense-like mechanism repurposed for tumor suppression.
Therapeutic potential for ICB sensitization: Targeting Fitm2 or GPI transamidase genes (Pigk, Gpaa1) enhances tumor regression in immunocompetent mice treated with anti-PD-1, but not in immunodeficient (NSG) mice, underscoring the immune-dependent nature of these dependencies.
Why should we care?
The key takeaway of this work is that cancer cells are not static targets, their survival relies on adaptive pathways activated by the immune microenvironment. By exploiting these context-specific weaknesses (e.g., with FITM2 inhibitors or drugs targeting GPI anchor biosynthesis), we might overcome resistance to immunotherapies like checkpoint blockade. However, the study’s preclinical nature and the lack of strong clinical correlations in human datasets temper enthusiasm, suggesting that while these pathways are promising, their therapeutic translation will require further validation. Critically, the work also highlights a double-edged sword: IFNs can both stimulate antitumor immunity (via antigen presentation) and promote tumor adaptation (via dependencies like FITM2 or GPI transamidase). This duality underscores the need for precision strategies that tip the balance toward immune-mediated tumor elimination rather than resistance
Biodiversity and biogeography of the multi-kingdom cancer microbiome
Dohlman et al., Cell (2026). 10.1016/j.cell.2026.04.015
The paper in one sentence
A rigorous pan-cancer analysis of 16,639 tumor genomes reveals that only orodigestive cancers harbor detectable, site-specific multi-kingdom microbiomes, which correlate with tumor mutation burden.
Summary
This study addresses long-standing controversies about the presence and distribution of microbes in human tumors by developing PathSeq-T2T, a robust host-subtraction and decontamination pipeline that leverages the complete T2T-CHM13 human reference genome. Applied to 16,639 high-depth tumor whole genomes from the UK 100,000 Genomes Project, the pipeline effectively removes human sequences and environmental contaminants. After decontamination, microbial signatures in most solid tumors were indistinguishable from background, resolving prior conflicting reports. However, orodigestive tumors (oropharyngeal, esophageal, gastric, and colorectal) consistently retained microbial signals, harboring polymicrobial, multi-kingdom communities, including bacteria, fungi, viruses, archaea, and even the protozoan parasite Trichomonas, that varied by tumor site, subtype, and genomic context. Notably, microbial load correlated with tumor mutation burden (TMB), particularly in hypermutated (MSI/POLE) subtypes, suggesting a link between microbial colonization and tumor genomic instability.
Personal highlights
Robust decontamination pipeline: PathSeq-T2T sets a new standard for low-biomass microbiome detection by combining T2T-CHM13 host subtraction, multi-classifier validation (Kraken2, MetaPhlAn4, Sylph), and a pan-cancer equiprevalence (PCE) score to distinguish true microbial signals from widespread contamination.
Most cancers lack a tumor microbiome: After rigorous decontamination, microbial signals in most solid tumors (e.g., brain, breast, lung) were indistinguishable from background, challenging prior claims of ubiquitous microbial colonization in cancer.
Orodigestive cancers are microbial hotspots: Colorectal, oropharyngeal, esophageal, and gastric tumors consistently harbored site-specific, polymicrobial communities, with microbial composition mirroring the biogeography of healthy tissues (e.g., Bacteroides in colorectal, Prevotella in oropharyngeal).
Multi-kingdom communities: Beyond bacteria, these tumors hosted fungi (Candida, Saccharomyces), archaea (Methanobrevibacter smithii), viruses (HPV, EBV), and, in rare cases, protozoa (Trichomonas), expanding the known diversity of tumor-associated microbes.
Link to tumor genetics: Microbial load correlated with tumor mutation burden (TMB), with hypermutated (MSI/POLE) subtypes showing 3.9–6.5-fold higher microbial density, suggesting a potential interplay between microbial colonization and genomic instability.
Why should we care?
This work provides a critical, methodologically rigorous resolution to the heated debate about the cancer microbiome. By addressing contamination, a major confounder in prior studies, it demonstrates that tumor-associated microbiomes are not universal but are largely restricted to cancers arising at mucosal barrier sites (e.g., the digestive and upper respiratory tracts), which are already colonized by microbes under normal conditions. The finding that microbial abundance scales with tumor mutation burden hints at a bidirectional relationship: while microbes may contribute to genomic instability (e.g., via genotoxins or immune modulation), hypermutated tumors might also create a more permissive niche for microbial growth.
Correcting spatial transcriptomics data affected by a prevalent transcript leakage problem across platforms, species, and tissues
Shi et al. bioRxiv (2026). 10.64898/2026.06.13.732076
The paper in one sentence
This study identifies and addresses a widespread transcript leakage problem in spatial transcriptomics (ST) data, where transcripts diffuse from their cell of origin to neighboring cells, by introducing DeLeakage, a reference-free Bayesian method that restores accurate gene expression and improves downstream analyses.
Summary
Spatial transcriptomics (ST) has revolutionized the study of tissue organization by mapping gene expression in its spatial context. However, Shi et al. reveal a systematic issue: transcripts often leak from their originating cells into nearby cells, leading to false detection of cell-type-specific markers in unexpected cell types and distorting spatial gene expression patterns. This problem is not platform- or tissue-specific, it affects imaging-based (e.g., MERFISH, Xenium) and sequencing-based (e.g., Pixel-seq) ST data across mouse brain, human heart, and other tissues. The authors propose DeLeakage, a Bayesian hierarchical model that decomposes observed transcript counts into endogenous expression and leaked transcripts, accounting for gene-specific diffusion properties and spatial neighborhood effects. Unlike existing deconvolution methods (e.g., SPLIT, SpotClean), DeLeakage does not rely on external references and models leakage as a distance-dependent diffusion process. The method is theoretically identifiable (proven in the paper) and computationally efficient, with both CPU and GPU implementations. Validation on simulated and real ST datasets shows that DeLeakage effectively removes leakage artifacts, improves cell-type annotation, and reduces false spatial expression signals.
Personal highlights
Transcript leakage is pervasive: Across multiple ST platforms (MERFISH, Xenium, Pixel-seq), tissues (mouse brain, human heart), and species, transcripts of cell-type-specific markers (e.g., Slc17a7 for excitatory neurons) are frequently detected in unrelated cell types, with spatial patterns suggesting diffusion from neighboring cells.
DeLeakage: A gene-specific, reference-free solution: The method models leakage as a distance-dependent diffusion process with gene-specific contamination parameters, allowing it to distinguish endogenous expression from leaked transcripts without requiring reference data (e.g., scRNA-seq).
Theoretical rigor: The authors prove model identifiability under realistic conditions, addressing a key limitation of prior deconvolution methods (e.g., non-identifiability in NMF-based approaches).
Outperforms existing tools: In benchmarks against SPLIT (reference-based deconvolution) and SpotClean (spot-swapping correction), DeLeakage more accurately restores true expression levels, reduces co-detection of mutually exclusive markers, and improves cell-type clustering (e.g., 71.7% increase in Adjusted Rand Index vs. 14.2% for SPLIT).
Scalable and practical: The GPU-accelerated implementation processes large ST datasets efficiently (e.g., 130K cells in ~1 hour), with lower memory usage than alternatives like SPLIT.
Why should we care?
This work highlights a prevalent but underappreciated issue in ST and provides a robust, theoretically grounded solution. For researchers using ST, it’s a call to re-evaluate past data and consider leakage correction as a standard practice.

Other papers that peeked my interest and were added to the purgatory of my “to read” pile
Hierarchical classification of immune cell transcriptomes at population-scale
Decoding common and rare noncoding variant effects across cellular and developmental contexts
Somatic variant detection in normal tissues from single-cell sequencing data
Benchmarking gene expression reconstruction from single-cell latent representations
Stromal Prostaglandin is a Dominant Spatial Regulator of Cell-fate Plasticity in Colorectal Cancer
CD4+ T cells impair tumor growth through IL-3 and TNF-dependent vascular damage
Thanks for reading.
Cheers,
Seb.