


Weekly reads 10/2/25
Week 3! This week, I focused particularly on new developments in bioinformatics, except for the Seq-Scope-X preprint, which appears to be the first sequencing-based spatial method to achieve the same resolution as imaging-based techniques—a fantastic advancement that I can only applaud!
Additionally, I keep coming across many interesting developments in the new AI hype surrounding foundation models. While it can sometimes be challenging to assess the actual improvements claimed in these preprints and papers, the field seems to be moving in the right direction. However, the need for standardized and unbiased benchmarks is becoming increasingly urgent.
Preprints/articles that i managed to read this week
RegFormer: A Next-Generation Single-Cell Foundation Model Integrating Gene Regulatory Hierarchies
Hu et al. (2025). RegFormer: A single-cell foundation model powered by gene regulatory hierarchies. bioRxiv. https://doi.org/10.1101/2025.01.24.634217
The paper in one sentence
RegFormer is a single-cell foundation model that integrates gene regulatory networks (GRNs) and Mamba Blocks to improve biological interpretability and predictive accuracy in single-cell RNA sequencing (scRNA-seq) analysis.
Summary
Single-cell RNA sequencing (scRNA-seq) has transformed our ability to study cellular heterogeneity, but analyzing these high-dimensional, sparse datasets remains a challenge. Traditional deep-learning models struggle to capture complex gene regulatory interactions and often rely on natural language processing (NLP)-based architectures, which are not inherently suited for unordered gene expression data.
RegFormer introduces a biologically aware framework that integrates gene regulatory hierarchies into its model structure. It incorporates a graph-based topological sorting algorithm that arranges genes according to regulatory influence and employs Mamba Blocks, an advanced neural network architecture optimized for high-dimensional and long-range dependencies. Additionally, RegFormer features a dual-embedding system that separately encodes gene identity and gene expression levels, enhancing interpretability.
By training on 22 million human cells and 50 million parameters, RegFormer outperforms existing models (e.g., scGPT, Geneformer, scFoundation, and scBERT) across various tasks, including cell annotation, gene regulatory network (GRN) inference, genetic perturbation prediction, and drug response modeling
Personal highlights
Integration of gene regulatory networks (GRNs) into deep earning: RegFormer leverages GRNs to impose biological constraints on how gene expression is modeled, aligning computational predictions with real-world regulatory relationships.
Graph-based topological sorting for gene order optimization: RegFormer introduces a novel topological sorting algorithm that orders genes based on their regulatory relationships, ensuring a biologically meaningful sequence for training.
Mamba blocks for efficient single-cell Modeling: These combine state-space models (SSMs), multi-layer perceptrons (MLPs), and convolutional layers to efficiently model long-range gene dependencies.
Dual-embedding strategy for enhanced biological interpretability:
Value embedding: Captures gene expression levels.
Token embedding: Captures gene identity and regulatory role.
RegFormer outperforms state-of-the-art models in cell annotation, GRN inference, perturbation modeling, and drug response prediction
Why should we care?
RegFormer provides a new computational framework that not only improves accuracy and scalability but also ensures biological interpretability—a critical step toward making machine learning models useful in real-world clinical and research applications. By incorporating gene regulatory hierarchies and employing next-generation AI techniques, RegFormer represents a major advancement in single-cell bioinformatics, bridging computational power with biological relevance.
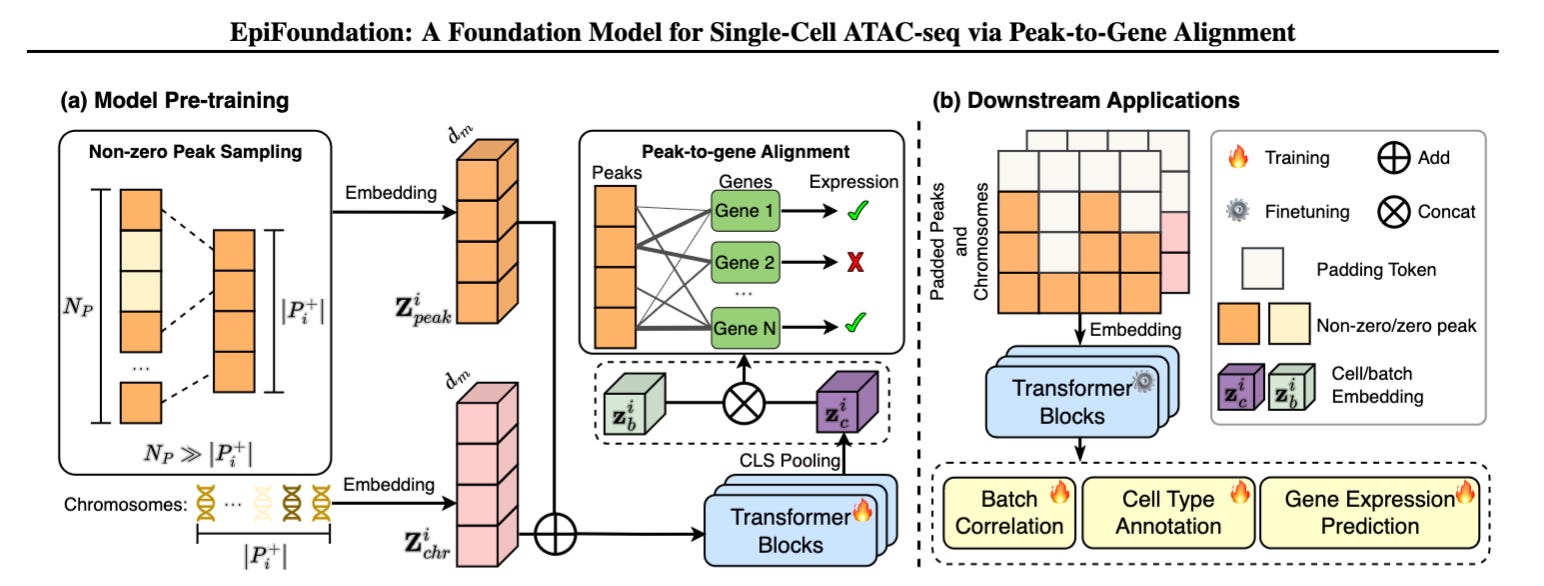
EpiFoundation: A Foundation Model for Single-Cell ATAC-seq with Peak-to-Gene Alignment
Wu et al. (2025). EpiFoundation: A Foundation Model for Single-Cell ATAC-seq via Peak-to-Gene Alignment. bioRxiv. https://doi.org/10.1101/2025.02.05.636688
The paper in one sentence
EpiFoundation is a single-cell ATAC-seq foundation model that overcomes data sparsity by leveraging non-zero peak representations and gene expression supervision to improve chromatin accessibility analysis and downstream regulatory predictions.
Summary
Single-cell ATAC-seq (scATAC-seq) enables researchers to study chromatin accessibility at single-cell resolution, providing insights into gene regulation. However, its high data sparsity and computational cost have limited the development of foundation models tailored to this data type. While existing scRNA-seq models (e.g., scGPT, Geneformer) use self-supervised learning, they do not effectively handle scATAC-seq data due to its high-dimensionality and binary nature.
EpiFoundation introduces two key technical innovations:
Non-zero peak representation – Instead of modeling the entire peak set, the model focuses only on accessible peaks, improving efficiency and enhancing the density of cell-specific information.
Peak-to-gene alignment supervision – By using paired scRNA-seq data as a guide, EpiFoundation aligns chromatin accessibility signals to gene expression, learning biologically meaningful regulatory relationships.
The model was pre-trained on 100,000+ cells from 19 tissues and 56 cell types (MiniAtlas dataset), demonstrating state-of-the-art performance in cell type annotation, batch correction, and gene expression prediction.
Personal highlights
Efficient representation of sparse scATAC-seq Data: Instead of processing all peaks, EpiFoundation models only non-zero peaks, significantly reducing computational overhead while maintaining biological relevance.
Cross-modality supervision with scRNA-seq: Gene expression data is used as a supervisory signal to align chromatin accessibility with transcriptional output, improving regulatory inference.
Transformer-based peak embeddings with chromosome context: Each peak is embedded alongside its chromosome identity, helping the model preserve genome structure while learning regulatory interactions.
Why should we care?
By tackling the challenges of data sparsity and regulatory complexity, this model improves our ability to connect epigenetic states with gene expression, which is crucial for understanding disease mechanisms, cellular identity, and transcriptional regulation. Moreover, its scalable and generalizable approach sets the stage for the development of multi-modal foundation models that integrate diverse single-cell data types.
Seq-Scope-X: Pushing the Boundaries of Spatial Transcriptomics Beyond Optical Resolution
Anacleto et al. (2025). Seq-Scope-eXpanded: Spatial omics beyond optical resolution. bioRxiv. https://doi.org/10.1101/2025.02.04.636355
The paper in one sentence
Seq-Scope-X integrates tissue expansion with high-resolution sequencing-based spatial transcriptomics, achieving sub-200 nm resolution and enabling multi-omics profiling at near-nanoscale precision.
Summary
Spatial transcriptomics (ST) has revolutionized our ability to map gene expression in intact tissues, but sequencing-based methods (sST) have been limited by resolution constraints due to RNA diffusion and sequencing pixel sizes. Seq-Scope-X overcomes this by incorporating tissue expansion techniques into submicrometer-resolution Seq-Scope, achieving spatial transcriptomics beyond the diffraction limit of optical microscopy (~200 nm). By physically enlarging tissues, Seq-Scope-X reduces transcript diffusion effects and increases spatial feature density by an order of magnitude. This advancement allows researchers to distinguish nuclear and cytoplasmic transcriptomes at the single-cell level, revealing subcellular transcriptomic heterogeneity. Seq-Scope-X also supports high-resolution spatial proteomics, demonstrating its multi-omics potential.
Personal highlights
Sub-200 nm resolution with tissue expansion: Seq-Scope-X surpasses diffraction-limited microscopy by physically enlarging tissue sections, achieving a 10× increase in spatial resolution compared to previous sST methods, Enabling the mapping of nuclear and cytoplasmic transcriptomes at near-nanoscale precision.
Simultaneous spatial transcriptomics and proteomics: Seq-Scope-X supports barcode-tagged antibody staining, enabling high-resolution spatial proteomic analysis alongside transcriptomics.
Despite its multi-step process, Seq-Scope-X maintains high RNA capture efficiency, comparable to standard sequencing-based ST methods (bulk RNA-seq: ~0.9 correlation).
Increased complexity & experimental cost: tissue expansion requires additional preparation steps, increasing the time and expertise required to perform experiments. The multi-step process could also introduce potential RNA loss or structural artifacts if not optimized correctly.
Higher nackground signal in spatial proteomics: Non-specific antibody binding in spatial proteomics introduces background noise, which could compromise signal specificity.
Optimization required for nanoscale expansion (<100 nm): While dimethylacrylamide (DMAA) expansion gels have been tested to push the resolution below 100 nm, achieving true nanoscale resolution remains a work in progress.
Why should we care?
Seq-Scope-X bridges the gap between sequencing-based and imaging-based spatial transcriptomics by combining whole-transcriptome coverage with super-resolution spatial mapping. Its ability to resolve subcellular transcriptomic heterogeneity makes it a powerful tool for studying cellular function, disease mechanisms, and tissue architecture at an unprecedented level of detail.
scGPT-spatial: Continual Pretraining of a Single-Cell Foundation Model for Spatial Transcriptomics
Wang et al. (2025). scGPT-spatial: Continual Pretraining of Single-Cell Foundation Model for Spatial Transcriptomics. bioRxiv. https://doi.org/10.1101/2025.02.05.636714
The paper in one sentence
scGPT-spatial extends the scGPT foundation model by incorporating spatially aware continual pretraining, integrating a Mixture-of-Experts (MoE) decoder and spatially aware sampling to improve the analysis of spatial transcriptomics data.
Summary
Existing foundation models, such as scGPT, have been pretrained on scRNA-seq data but lack spatial awareness and fail to capture multi-modal differences across sequencing platforms. scGPT-spatial builds upon scGPT through continual pretraining on SpatialHuman30M, a dataset of 30 million spatial transcriptomics profiles from sequencing- and imaging-based protocols. It introduces two major innovations:
A Mixture-of-Experts (MoE) decoder, which adaptively routes data based on sequencing modality, ensuring accurate protocol-aware gene expression predictions.
Spatially aware sampling, which groups cells based on proximity, enabling the model to capture microenvironmental effects and cell-cell interactions within tissues.
These innovations allow scGPT-spatial to outperform existing methods in cell type clustering, multi-slide integration, gene imputation, and cell-type deconvolution while offering a unified foundation for spatial transcriptomics analysis.
Personal highlights
Continual pretraining on 30 million spatial profiles: scGPT-spatial is trained on four major sequencing modalities (Visium, Visium HD, Xenium, MERFISH), enabling it to generalize across diverse datasets.
Mixture-of-Experts (MoE) decoder for protocol-specific adaptation: this decoder learns sequencing protocol-specific biases, ensuring more accurate and modality-aware gene expression predictions.
Spatially aware sampling and training strategy: Unlike traditional single-cell models, scGPT-spatial incorporates spatial proximity into training, learning cell-cell interactions within tissue architecture.
scGPT-spatial surpasses existing models in cell-type clustering, multi-slide batch correction, gene expression imputation, and cell-type deconvolution.
Zero-shot generalization across datasets: Without additional fine-tuning, the model effectively integrates multi-modal and multi-slide datasets, making it adaptable for various biological applications.
Why should we care?
scGPT-spatial represents a significant breakthrough in spatial transcriptomics analysis, providing a generalizable, multi-modal foundation model that integrates sequencing- and imaging-based data while improving biological accuracy in cell-type and tissue mapping. As spatial omics continues to expand, models like scGPT-spatial will enable deeper insights into tissue organization, disease mechanisms, and therapeutic target discovery

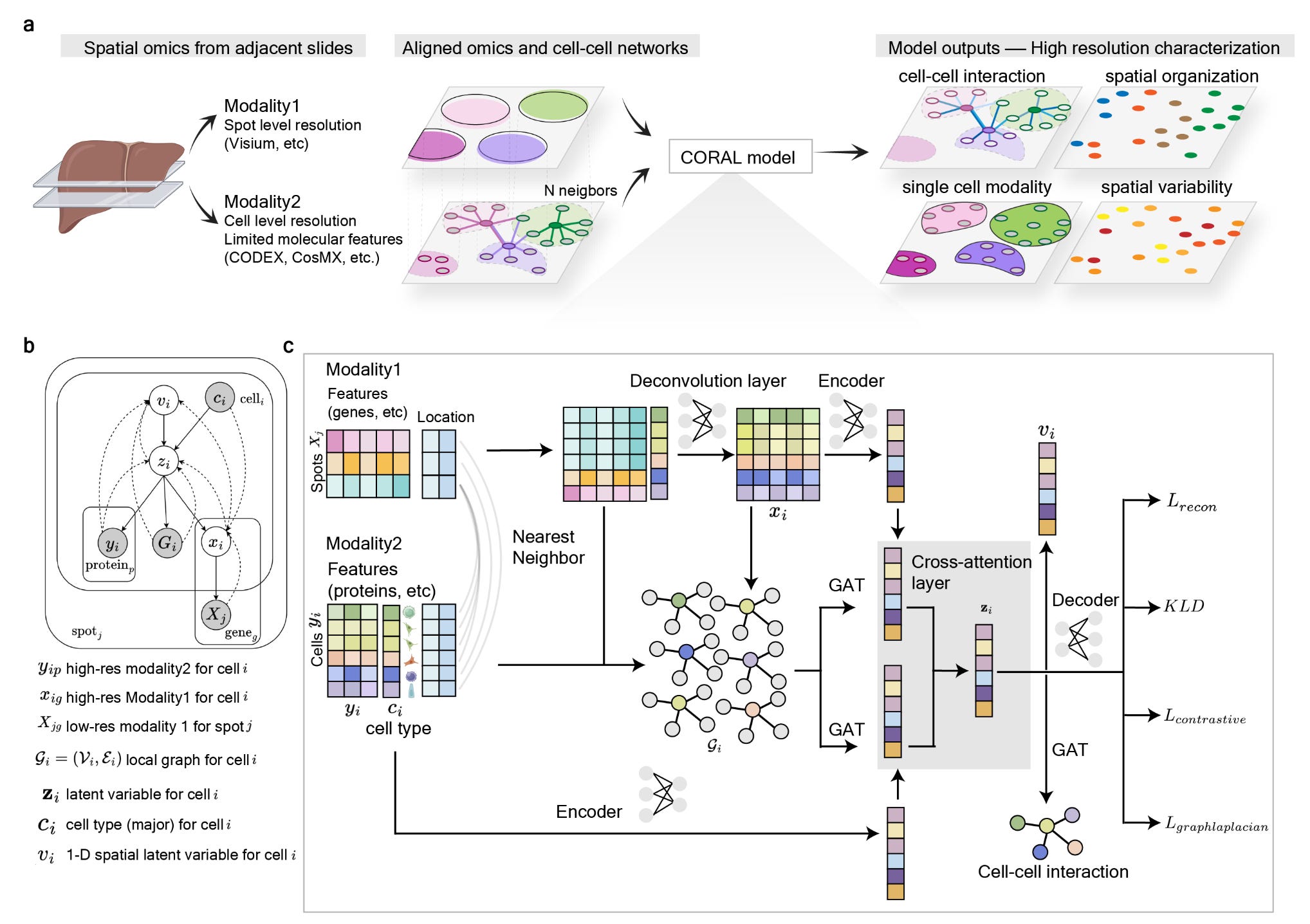
Unlocking Spatial Multiomics: CORAL’s Graph-Attention Approach to Tissue Organization
He et al. (2025). Learning Single-Cell Spatial Context Through Integrated Spatial Multiomics with CORAL. bioRxiv. https://doi.org/10.1101/2025.02.01.636038
The paper in one sentence
CORAL is a probabilistic deep generative model that integrates different spatial modalities using graph attention networks, enabling high-resolution tissue analysis at the single-cell level.
Summary
The CORAL framework addresses a major challenge in spatial omics: integrating data from different molecular profiling techniques that vary in resolution and coverage. By leveraging graph-based attention mechanisms, CORAL harmonizes multimodal spatial data, deconvolutes lower-resolution spot-based data into single-cell resolution, and uncovers cell-cell interactions within complex tissue architectures.This model provides a more accurate and interpretable representation of cellular microenvironments.
Personal highlights
Graph-Attention networks for spatial relationships: CORAL models interactions between neighboring cells, allowing precise inference of cellular communication networks.
Multimodal integration across resolutions: The model aligns data from high-resolution single-cell proteomics (e.g., CODEX) and lower-resolution spatial transcriptomics (e.g., Visium), providing a unified molecular landscape.
Single-cell deconvolution from spot-level data: CORAL infers gene expression at single-cell resolution from bulk spatial transcriptomic signals, significantly improving data granularity.
Probabilistic deep generative model: The framework utilizes a variational inference approach to capture biological variability and improve prediction accuracy.
Contrastive learning for cell-Type resolution: CORAL enhances cell-type classification by encouraging distinct molecular signatures for different cell populations while preserving similarities within the same type.
Why should we care?
By resolving single-cell heterogeneity and refining spatial multiomics integration, CORAL paves the way for deeper insights into disease mechanisms, for example in oncology and immunotherapy.

Other papers that peeked my interest and were added to the purgatory of my “to read” pile
Oncofetal reprogramming drives phenotypic plasticity in WNT-dependent colorectal cancer: https://www.nature.com/articles/s41588-024-02058-1
Sex Differences in Cancer Functional Genomics: Gene Dependency and Drug Sensitivity: https://www.biorxiv.org/content/10.1101/2025.02.05.636540v1
Principled PCA separates signal from noise in omics count data: https://www.biorxiv.org/content/10.1101/2025.02.03.636129v1
Thanks for reading.
Cheers,
Seb.